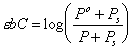- 3.5 Ultraviolet and Visible Absorption Spectroscopy
Molecules in solution have continuous absorption spectra in the UV and visible region. Absorbance arises from transitions in the valence electrons of molecules and is affected by such things as solvation and molecular interactions. Figure .10 shows an example of a spectrum of an organic molecule, fluoranthene. Changes in oxidation states of species have large effects on absorption spectra. Because the absorption peaks are relatively broad, UV/Vis spectrometry is most frequently used for quantitative analysis. It is also widely used in liquid chromatography detectors. A schematic diagram of a UV-Vis spectrophotometer is shown in Figure .11.
Quantitative analysis is performed by determining the absorptivity coefficient, e, using solutions of known concentrations. A wavelength best suited for analysis is chosen. This is usually selected at a place in the spectrum where the absorbance is not changing rapidly, and where interfering substances have low absorbance. While it is not necessary to select a point at which the absorbance is at a maximum, selecting the highest absorbance point will give the highest sensitivity. If the samples to be analyzed are not at trace levels, a wavelength at which lower absorbance takes place may be preferable. The best accuracy in absorbance spectroscopy is achieved at fairly low absorbances. The point of best accuracy is at an absorbance of 0.38. At higher concentrations, because of the log term in Beer's law, a large change in concentration causes only a small change in the transmitted light. Therefore it is better to use a wavelength at which absorbance is less, if it avoids measuring solutions with absorbances above 1, or diluting the sample.
Several analytes can be determined simultaneously in the same solution, because of the additivity of Beer’s law. Wavelengths are selected where the differences in the extinction coefficients are as large as possible, to minimize error. For instance, if one is determining a mixture of A and B, the absorption spectra of the pure compounds should be examined to find a point at which A absorbs strongly and B minimally, and another point where B is absorbing strongly, and the absorbance of A is low. The extinction coefficients for each component at each selected wavelength are determined from standards. The cell path length, b, is constant. Absorbance measurements are then made at different wavelengths, producing set of simultaneous equations with only the concentrations as unknowns. One wavelength measurement is required for each component, to give one equation for each unknown concentration. In practice, the method is limited to two or three components, as the errors of measurement rapidly increase when components are added.
- UV and Visible Instrumentation
-
Light Sources
Tungsten filament lamps are common broad band sources used in the visible region of the spectrum. They have an operating temperature of about 3000o K. At this temperature the peak of radiation is actually in the near infrared, and the emission in the visible range is only a small fraction of the total energy emitted from the lamp. While running the lamp filament at a much higher temperature will give more radiant energy in the visible, it will also shorten the lamp's useful lifetime. The use of bromine or iodine vapor in the lamp fill gas, combined with a fused silica envelope allows a longer lifetime at elevated temperatures. So these quartz-halogen lamps are widely used. Xenon arc lamps require higher voltages but are often used as excitation sources for fluorescence measurements, because they produce a wide continuum, which extends into the ultraviolet region. In the ultraviolet, hydrogen or deuterium electrical discharge lamps are used. These lamps are filled with a low pressure of hydrogen or deuterium, and a DC voltage of about 40 volts is applied. The envelope is made of quartz or fused silica. The low wavelength cutoff of these lamps depends on the transmission of the window material, and is usually about 180 - 200 nm.
- UV-Vis Detectors
Photoemissive vacuum tubes are useful in the range from 120 to about 1000 nm. The tubes are composed of a photocathode which will emit electrons when light falls upon it. The electrons are collected on an anode, producing an electrical current. Various combinations of photocathode materials and window materials make different tubes suitable for different ranges of wavelengths. Because of the low currents generated by low radiation levels, amplification may be necessary. These tubes have a dark current that flows when there is no incident radiation, so the lowest measurable currents must exceed the dark current by a significant amount. Amplification cannot extract signals which are not significant with respect to the dark current because both the signal and the dark currents are amplified to the same extent.
Photomultiplier tubes are considerably more sensitive because these, as their name implies, multiply the effect of each photon that falls on the detector. They are composed of a photoemissive cathode and a series of dynodes at successively higher voltages. The incident photon ejects an electron from the cathode. This falls on the first dynode which emits several electrons in response. These are focused onto the next dynode, where the same process is repeated. Thus, the response to each photon is multiplied by the number of electrons emitted for each incident electron (f) and by the number of stages (n). The overall multiplication of signal, or gain (G), of the photomultiplier is G = f n. The value of f depends not only on the material of the dynodes but also on the potential difference imposed on these dynodes. Therefore, the sensitivity can be controlled over a wide range by adjusting the potential on the multiplier tube. Because of their sensitivity, these are used only for detection of low light levels. A photomultiplier, shown in Figure .12, should be protected from light as much as possible when it is being handled or mounted into equipment, even when it is not powered.
Photodiodes are solid state devices. In these silicon-based devices, electrons are promoted to the conduction band when they are irradiated with light in the visible or near infrared areas of the spectrum. These give a response that is larger than that of photoemissive tubes, but very much less than that of photomultipliers. Since they are solid state devices, they can be made very tiny. This allows a row of them to be placed so that each reads a narrow wavelength band, at the same time. From the output of such a diode array detector an absorption or emission spectrum can be obtained instantly, without the need for scanning the spectrum across a single detector. This is both more rapid and less expensive to implement, since scanning requires expensive precision moving parts.
- Ultraviolet - Visible Spectroscopy Samples
Samples used in ultraviolet and visible spectroscopy are usually in solution form, although gas phase measurements can be made using long path gas-tight cells. The sample cells are made of quartz for UV analysis and of glass or plastic for use in the visible region. Round test tubes are often used for low precision work. It is difficult to get a reproducible path length in a round tube, because of slight differences in the placement of the tube in the light beam. It is common practice to mark the tube so that it can be replaced in the spectrometer in the same orientation each time. The tubes used to hold samples and standards can be matched by filling them with an absorbing solution, and selecting the tubes which give the same absorption readings. For more accurate work, cuvets which have flat parallel sides are used. These are also available in various materials, and are considerably more expensive than test tubes. Cuvets are made in various path lengths, with a common size being 10 mm.
The samples and standards must be free of particles and the cuvets should be carefully wiped to avoid fingerprints on the surface. These impurities can scatter light and reduce the transmission of light through the sample. The calibration standards should be held in matching cuvets and a solvent blank should also be prepared for zeroing the instrument.
Gas phase UV measurements are useful for stack monitoring purposes. A non-dispersive technique is most commonly used for monitoring such gases as SO2, NO2, CO2. A filter system is used rather than a monochromator. An optimal wavelength for the analyte is selected, as well as a background wavelength. For SO2, these are 280 nm for the analyte and 578 as the background. The filters are mechanically rotated through the light path so that the absorbance is measured at the analytical wavelength and the background wavelength alternately. The sample is drawn through a long path gas-tight cell, where its absorbance is measured continuously. This apparatus is shown in Figure .13.
- Colorimetry
To determine individual compounds in samples, a reagent which reacts specifically with the component of interest is added to the sample, forming a colored species. The intensity of color produced is proportional to the concentration of analyte in the sample and can be measured using the UV-visible spectrophotometer.
Analyte + Colorimetric reagent ®
Colored complex ( .16)
The selectivity is provided by the colorimetric reaction and the absorbance in the visible or UV region is used only for quantitation.
Many colorimetric reagents are available for specific metal ions as well as for organic pollutants. Some of these are listed in Table .2. A major advantage of this type of measurement is its simplicity. For example, to measure the amount of Cr (VI) in a water sample, the pH of the sample is adjusted to around 2 by adding H2SO4. A few ml of a 1,5-diphenyl carbazide solution is added to the sample. In five to ten minutes, a pink color is formed, whose intensity can be measured at 540 nm using a UV-Vis spectrophotometer. Similarly, Cr(III) can be determined by first oxidizing it to Cr(VI) by boiling with KMnO4 and continuing as above.
Another example of a colorimetric test is the measurement of NO3- in soil. Nitrates are extracted from a soil sample with a 0.01 M CuSO4 solution containing Ag2SO4 . The latter prevents interference from chloride. The extract is treated with phenoldisulfonic acid. The colorimetric reaction depends upon nitration of position 6 of 2,4-phenoldisulfonic acid:
C6H3OH(HSO3)2 + HNO3 ®
C6H2OH(HSO3)2NO2 + H2O ( .17)
In alkaline solution, the product is yellow in color.
The selectivity and sensitivity of these methods are often not as good as other methods. For example, atomic absorption usually provides interference-free measurement of metals at lower detection limits than the colorimetric methods. The main advantage of colorimetry is that the analysis can be done using a simple, inexpensive spectrophotometer. The availability of battery powered spectrophotometers make this technique easily adaptable to field use. While colorimetric tests are usually designed to be specific for a selected analyte, they are prone to interferences. Detection limits depend upon the analyte/colorimetric system, but measurements down to ng/g levels are possible.
Colorimetry can be used for air, water, soil and biological samples. In the case of air analysis, the air is usually drawn through an chemical reagent to trap the target pollutant. Then, a colorimetric reagent is added to produce the color. In water analysis, the colorimetric reagent may be added directly to the water. In soil (or solid) samples, the soil is first extracted and the colorimetric reagent is added to the extract.
- 3.6 Infrared Spectroscopy
Infrared spectroscopy has found its major use in environmental analysis in the field of long range sensing of air pollutants. To obtain the necessary sensitivity, the path length of the sample is lengthened. Long path cells can be used. Open path measurements where the analyzing beam is reflected from a remote mirror to a telescopic detection apparatus are used in the field. The air through which the beam travels is the sample.
The infrared absorption spectrum of a compound is governed by the absorbance of energy corresponding the energy transitions in the vibrational and rotational modes of the molecule. Each bond in a molecule has a vibrational frequency which depends on the mass of the two atoms connected by the bond and by the strength of the bond. Therefore, certain frequencies are indicative of certain bonds. Table .3 shows the characteristic absorption wavelengths for some functional groups. For instance a peak found at 2960-2870 cm-1 is usually due to the absorbance by the methyl group. Other frequencies indicate certain functional groups such as aromatic rings, OH groups, NH groups, etc. Absorbance peaks correlated with the presence of certain functional groups can be found in any reference work on IR. Figure .14 shows the spectra of benzene and trichlorobenzene. The difference in the absorbance in the C-H stretching band near 3100 cm-1 shows that the chlorinated benzene has fewer C-H bonds, while the appearance of peaks in the region of 650 to 750 indicate the presence of C-Cl bonds. The environmental analyst usually uses spectra to identify molecules by matching their spectra with library spectra, or to confirm the identity of a molecule which is thought to be present. The IR spectrum is not often used in environmental work to deduce the structure of compounds which do not yet appear in spectral libraries, as is frequently done in synthetic organic chemistry. Therefore, we will not discuss the interpretation of spectra in detail.
IR absorption obeys Beer's law and can be used to determine concentration of the absorbing species. IR spectra are usually presented as a plot of percent transmission versus wavenumber. More modern instruments with computerized data handling can also display an absorbance spectrum. However the data are presented, the difference between the baseline and the peak absorbance (not transmission) must be measured before a concentration/absorbance calibration may be made.
- Scanning Infrared Instrumentation
-
IR Sources
In the infrared region the common sources are electrically heated elements made of ceramic or alloys. The Nernst glower is composed of rare earth oxides, operates up to about 1800oK, and has a negative coefficient of electrical resistance. This means that the resistance becomes lower as the source is heated, and it may require preheating before a current can be passed at all. The globar is a silicon carbide rod, which operates at a lower temperature, about 1600oK, and gives more radiation in the region below 1500 cm-1 than does the Nernst glower. A characteristic of all the IR sources is their generally low output of radiation. This means that IR spectroscopy is generally energy limited, and requires sensitive detection.
- Infrared Monochromators
Since infrared radiation will not pass through glass or quartz optics, the monochromators are constructed using reflective gratings and front surface curved mirrors to diffract and focus the radiation. The range of wavelengths covered is too large to be diffracted efficiently by a single grating, so the instrument usually contains several gratings supported on a rotating post. The grating is rotated slowly when the sample is being scanned, then the scan is halted, and another grating is turned into place to scan the next region of the spectrum.
- Fourier Transform Infrared Spectrometry (FTIR)
The inherent sensitivity of IR spectroscopy is low, due to the limitation of the energy available from the source and the low sensitivity of the IR detectors. Therefore, a design using a Michaelson interferometer instead of a monochromator is often used. This is the basis of Fourier Transform Infrared (FTIR). To understand the functioning of an FTIR, one has to understand time domain spectroscopy.
Conventional spectroscopy involves measurements in the frequency domain, i.e., radiant power (expressed as absorbance or transmittance) is measured as a function of frequency. In time domain spectroscopy, radiant power is measured as a function of time. However, both these measurements contain the same information. Conversion between time and frequency domain can be achieved using a mathematical technique called Fourier Transform. The conversion between time and frequency domain is shown in Figure .15. If signal intensity is plotted as a function of time a time domain spectrum is produced. The frequency of this signal, when plotted, yields a single line, indicating that only one frequency was present in this signal. This is the frequency domain signal. When, multiple frequencies are present, the spectrum becomes more complex. This is shown in Figure .16, where the time domain spectra contains two frequencies. The A plot shows the two out-of -phase waves, and B shows their sum. The frequency domain plot shows that only two frequencies were present.
In FTIR, the measurements are done in the time domain rather than in frequency domain. A Michelson interferometer is used for this purpose. The interferometer is shown in Figure .17. There are two mirrors, one fixed, the other movable. The moveable mirror travels at a constant velocity. Radiation from the source is passed through a beam splitter, so that half of the beam reaches the movable mirror while the other half is reflected from the fixed mirror. The reflected beams from the two mirrors recombine at the beam splitter. Constructive and destructive interference takes place, depending upon the difference in path length between the path followed by the beam reflecting from the fixed and that from the movable mirror. This interference pattern is seen at the detector. For example, assume that the incident beam is a monochromatic beam. As the movable mirror moves forward, constructive and destructive interference occurs alternately. The resulting detector signal is a sine wave as shown in Figure .18. In other words, the interferometer converted the frequency domain spectra (the monochromatic light) into a time domain spectra. When many frequencies are present, the detector output is as shown in the second plot. The time domain spectra from the interferometer is called an interferogram. When the Fourier transform is performed on the interferogram, the frequency domain spectrum is constructed. The sample is placed in front of the detector, and some of the frequencies are absorbed by it. Fourier transform is then used to obtain the IR spectrum from the interferogram.
- Advantages of FTIR
IR sources have low intensity, and, when a monochromator is used, the IR beam is further attenuated by the slit. The FTIR design eliminates the need for a monochromator, so there is no attenuation of the IR beam. Therefore, the power of the beam reaching the detector is significantly higher and a high signal to noise ratio is obtained. This is referred to the throughput or Jaquinot advantage.
In a scanning instrument, it takes several minutes to scan the whole infra red region. In FTIR, there is no scanning to be done, and the spectrum of the whole region can be obtained in a second or less. FTIR has high wavelength accuracy and precision allowing many scans to be taken and their signals averaged. The signal has a fixed pattern, and the noise is random in nature. When scans are averaged, the signal increases while the noise is canceled out. The enhancement in signal to noise ratio is proportional to the square root of the number of scans averaged. For example, Figure .19 shows the result from a single rather noisy scan, and that obtained when 100 scans are averaged. While it may take 1500 seconds to obtain a spectrum in a scanning IR, the same spectrum can be obtained by an FTIR in about 1 second. Therefore, in 1500 seconds, 1500 scans could be obtained and signal averaged, which would enhance the signal to noise ratio by a factor of the square root of 1500 or 39.
The Fourier transform infrared spectrometer (FTIR) depends heavily upon computer power to deconvolute the interferogram and reconstruct it into a spectrum. As computers have become more powerful and inexpensive, the cost difference between the FTIR and the grating IR instrument has steadily diminished. Since the FTIR can report an IR spectrum in a few seconds, compared to several minutes for a grating instrument, the small additional cost for the FTIR is usually justified. Resolution is also better than in comparable grating instruments. Finally, because the entire spectrum is produced in digital form in the computer, subsequent data analysis is easily done. Backgrounds can be subtracted, and a spectrum can be compared to thousands of standard spectra in a computer library, with a few keystrokes.
- Samples for Infrared Spectroscopy
Because of the low power of IR sources, most solid or liquid IR samples must be rather thin. Gaseous samples, being of low concentration, need a longer path length. A common application of IR spectroscopy in environmental analysis is for long path sensing of pollutants in air. Gases can be placed in a cylindrical cell with IR transparent windows at the ends. The cells have inlets and outlets so that they can be purged with the sample gas. The windows are cut from a sodium chloride crystal or other IR transparent material. The pressure of the gas sample can be varied. Highly absorbing samples can be measured at pressures of a few millimeters of mercury, while low concentration samples can be used at higher pressures. For trace level samples, the required path length may be so long that the cell is impossibly bulky. In this case the light path may be folded, by placing front surface mirrors inside the cell. These are aligned so that the incoming beam of radiation is bounced back and forth through the sample several times before being brought to the exit slit. The adjustment and alignment of these cells are usually carried out using a visible laser beam. Figure .20 shows a long path cell.
For air pollution studies it is sometimes easier to bring the instrument to the sample. Long range IR analysis is done using an IR source beam which is passed over a path many meters in length. Figure .21 shows such a system. A gold plated reflector is mounted some distance away from the source and detector. The reflectors are gold plated mirrors, formed from a series of corner cubes. This arrangement of mirrors, lining the inside surface of three planes meeting at right angles, will always direct a beam that falls into it back along the same path as the incident ray. The beams are thus directed back to the source where they are intercepted and fed into an interferometer to be analyzed. The system is calibrated by using a gas cell of 10-15 cm path, filled with a known concentration of a standard gas mixture, and placed in the beam. The path length of the cell and the known distance to the reflector are used in the calculations.
The difficulties with the open long path systems are due to changes in high level components of the air, such as water vapor and carbon dioxide. This causes large changes in the IR spectrum at certain wavelengths. The complex mixture of organic compounds in the air also makes it difficult to determine specific compounds in the sample. However, if a single component is present at high concentrations compared to other possible absorbers at a particular wavelength, it is possible to follow the changes in that compound. This has been used to watch the components of automobile emissions over a roadway, and the results showed good correlation with the traffic density. It is also useful for industrial fenceline monitoring, where the compounds likely to be emitted are well known. Mathematical models involving the specific absorbances of compounds at different wavelengths are being used to develop algorithms to find the signature of specific pollutants in the mixture. This is possible in IR spectroscopy because of the information richness of the spectra. Another concern is that the concentration is integrated over the length of the path. A 500 m path system will show the same results if the entire air mass has a concentration of target compound of 1 ml/m3 or if a plume 5 m wide crosses the beam, carrying a concentration of 100 ml/m3.
Solid samples for IR analysis are handled by grinding the samples finely and mixing with refined mineral oil. This mull is smeared onto a transparent plate such as a sodium chloride plate. The absorbance spectrum is then taken. This technique is suitable for qualitative analysis, but is not suitable for quantitative studies because the sample thickness is not easily determined. Samples may also be ground finely with potassium bromide powder, and pressed into a pellet in a die. High pressure dies in which the samples are pressed while under vacuum can be used to form pellets which have little adsorbed water. Cells which have known spacings of a millimeter or so between the plates are available for liquid samples.
Quantitation is difficult in IR spectroscopy of liquids and solids primarily because setting the zero and 100% transmittance points is not readily done. The zero point, because of the low amount of energy reaching the detector, is difficult to set reproducibly. The 100% transmittance point cannot usually be set using a matched cell filled with the solvent because matched IR cells are not generally available. Therefore, a baseline method is usually used. The baseline is determined from the points of maximum transmission on either side of the peak of interest. The difference between the minimum of transmission and the zero point is considered I. The difference between the baseline and the zero point is Io. Then the absorbance can be calculated as:
A = - log ( I / Io ) ( .18)
Because of the relatively wide band passes used in IR methods, deviations from Beers law are common. Quantitation by IR spectrometry is generally not as precise or accurate as that done by UV or visible spectrometry. However, it has a particular capability of measuring a particular functional group in a complex mixture. This allows the estimation of, for instance, total ketone content or total aromatic content of a mixture, without separation.
In gas phase work, the cell path length is much greater and much easier to measure accurately. Therefore quantitative work in gases is more reliable and precise, if interferences can be avoided and if suitable standards can be obtained.
Example: An open path IR system is set up to study the concentration of SF6 in the air. The SF6 is to be used in a tracer study to see how gases emitted from a pollution source will be dispersed in the area under different weather conditions. The IR system is calibrated using a standard gas mixture containing 10 ml/m3 of SF6 in a 15.0 cm cell. The peak absorption occurs at about 945 cm-1. The reflector is placed 18.0 m from the source/detector apparatus. The absorbance measured in the standard gas is 0.35. The absorbance measured in the atmosphere is 0.22. Assuming that Beers law is obeyed, calculate the concentration of SF6 in the atmosphere during the measurement time. (The calculated value will be averaged over the time of collection and over the distance between the mirror and the detector.)
The absorbances measured are assumed to follow Beer's law, A = abC. The extinction coefficient for the SF6 is calculated from the standard as:
0.35 = a (0.15 M) (10 ml/m3)
a = 0.23
The path length for the open path system is 36 M, because the beam travels to the mirror and returns.
0.22 = (0.23) (36 M) C
C = 0.027 ml/m3 in the air.
Question: Referring to a table of characteristic IR absorption bands, what interferences do you think should be considered, and how would you attempt to compensate or correct for these?
-
3.7 Atomic Absorption Spectroscopy
For elemental analysis, especially for the determination of metals, it is often preferable to decompose the sample molecules into atoms and to measure the absorption or emission of radiant energy due to these atoms. The advantage of these methods is that atomic spectra are line spectra, and do not include broad absorption and emission bands. This makes it easier to select individual elements from a complex mixture, with much less chance of interference.
Atomic absorption (AA) is mainly used for analysis of metals in air, water and solid samples. The absorption spectra from AA is composed of narrow lines about 0.2 -0.4 nm wide. Even the best monochromator does not produce such narrow bands. Consequently, it is not possible to select these bands from a continuous source by use of a monochromator. If the incident beam is wider than the absorption band, only a small fraction of the radiation is actually absorbed. Figure .22 shows the situation when a wide bandpass of light is being absorbed by a species which can only absorb a narrow segment of the light. The difference between no absorption and very strong absorption, or between low concentration and high concentration is very small, leading to very poor sensitivity. In addition, when the incident beam has a larger band width than the absorption band, deviation from Beer’s law may occur.
This problem is overcome by using a line source which has a bandwidth similar to that of the absorbing species. The figure shows that a larger fraction of the radiation available can be absorbed by the species being measured, if the concentration is high enough.
For this reason, hollow cathode lamps, line sources, are used as the radiation source in atomic absorption spectroscopy. These contain a cathode filled with the element to be used in creating the line emission, and a wire anode. Figure .23 shows a diagram of a hollow cathode lamp. The body of the lamp is filled with a low pressure of an inert gas, usually neon or argon. When a voltage of 300-400 volts is supplied to the electrodes, the fill gas atoms are ionized and fall into the cathode, sputtering off the element atoms. These atoms are in an excited state and, as they fall back to the ground state, they emit the characteristic wavelengths of the particular element's spectrum. The width of these lines depends on the amperage passing through the lamp. If the current is too high a dense cloud of atoms is produced, raising the pressure in the lamp. This causes many intermolecular collisions, which spread out the energies of the atoms and thus increase the bandwidth of the line. In addition, the dense cloud will contain some ground state atoms, and these will absorb some of the emitted light before it exits through the window, lowering the output of radiation. Self absorption produces a broadened band with lower intensity in the middle of the band and higher emission at the sides.
The requirement for a specific lamp for each element usually limits atomic absorbance work to the analysis of one element at a time. Some instruments are capable of doing up to four elements simultaneously, by using multiple lamps and detectors, passing the beams through the same flame simultaneously.
- Flame Atomic Absorbance Spectroscopy
The atomic absorption spectrometer requires that the sample be atomized, broken down into individual atoms, before it is passed into the radiation beam for absorbance measurement. In flame AA, a liquid solution containing the sample is aspirated into a flame. This is achieved using a nebulizer, which mixes the sample solution with gaseous fuel and oxidant to form a uniformly mixed aerosol of the solution. There are several different phenomena which take place in the flame while the measurement is occurring. Each drop first dries to a small salt particle, then evaporates completely. The ion clusters heat further until they absorb enough energy to dissociate into free atoms in vapor state. The beam is passed through the flame and absorbance by the atomized species in the flame is measured. It should be noted that the absorbance is proportional to the concentration of ground state atoms in the flame.
The flame provides a complex and reactive atmosphere. Metal atoms can undergo chemical changes, forming, for example, refractory oxides or hydroxides. Atoms can also lose electrons to form ions. Any process which converts free ground state atoms to other forms lowers the sensitivity because the ground state atoms are the absorbing species.
Figure .24 shows a typical AA flame apparatus. The burner usually has a long narrow slot from which the flame emerges, and the light beam passes along the length of the slot. This allows for a longer absorbing path length, and better sensitivity. A commonly used flame is fueled with acetylene, with air for an oxidizer. When a higher temperature is needed, or where excess oxygen must be avoided, other flame gases are used. Table .4 shows some of the more common flame gas combinations and the reasons for their use.
Because the atomic absorption measurements in the flame are done in a dynamic system, it is especially important to be sure that the samples and standards are in a similar matrix. The viscosity of the solutions, the behavior of the mist in the flame, its drying and evaporation characteristics, and even the droplet size, can all have an effect on the rate of formation of atoms in the flame. In usual work, all standards and samples are made up in dilute acidic aqueous solutions. Where the characteristics of the sample are such that the standards may not be made in a similar matrix, the method of standard additions is often used.
- Graphite Furnace Atomic Absorption Spectrometry
The electrothermal furnace, an alternative to flame atomization, may be used to atomize a sample for atomic absorption spectroscopy. This technique, often called the graphite furnace method, minimizes sample preparation, because both liquid and solid samples may be used. The weighed or measured portion of sample is placed in a small graphite tube which is held between two electrodes. Some furnace tubes are manufactured with a small sample stage inside the tube, called a L’vov platform, which helps to insure that the sample is atomized evenly. These systems have several different configurations but most allow the sample to be injected into the middle of a horizontal graphite tube. A current is passed through the walls of the tube, usually increasing the temperature in a programmed fashion. The initial stage heats the furnace to a fairly low temperature, usually just above the boiling point of the solvent, in order to dry the sample. Then the temperature is raised to a point at which the sample is ashed, destroying any organic material present. This temperature varies widely, depending upon the character of the sample matrix and the target metal.
Finally the current is increased in a sharp pulse to volatilize the metals into the light beam. This current pulse raises the temperature rapidly to over 2000oC, in a matter of a few seconds. A puff of atomic vapor is produced, and its absorbance is measured. The signal is in the form of a peak, as the concentration increases and dies away. The graphite furnace is diagrammed in Figure .25. Because the entire amount of sample is atomized and is measured immediately, the sensitivity of the method is usually higher than that obtainable with flame atomization. Also, the amount of sample needed is smaller than that needed for flame AA. A liquid sample of 2 to 100 m
l is usually injected, using a syringe. Small amounts of solids may be weighed directly into the furnace. However, it is imperative that the solid is be dried and ashed at a low enough temperature so the targeted metal is not vaporized. When samples can be atomized directly, a great deal of sample preparation can be eliminated. For instance, there are screening methods for lead in blood which require only a few microliters of sample injected directly into the graphite furnace for analysis.
There is a difference between flame atomized and furnace atomized samples. The flame aspirates a continuous flow of sample solution, keeping a constant concentration of absorbing atoms, for as long as is necessary to establish and note the absorbance. The furnace provides a puff of atoms in a small cloud, which is transitory. The concentration of the atoms increases then decreases, and the instrument must be designed to follow this signal rapidly and accurately. Modern atomic absorption spectrometers have microprocessor controlled signal acquisition circuitry which handles the recording and integration of these rapidly changing signals readily. Since the entire sample is atomized at once, and there is no rapid flow of gases to dilute the atom cloud, as there is in a flame, the residence time of the atoms in the light beam is much longer than in flame AA and the detection limits are usually lower.
The graphite furnace method is subject to matrix problems, as often happens with very sensitive methods. Some salts are quite volatile. For instance, lead chloride is more volatile than other salts of lead. Therefore lead in chloride-bearing samples may give a low value, because some of the lead is lost as chloride during the ashing and drying cycles. This problem may be addressed by addition of ammonium nitrate, which helps to release the chloride as ammonium chloride, before the lead is volatilized. Many other of these matrix modifiers are useful in specific cases. It is always advisable to match the matrix of the samples and standards as closely as possible, and also to perform recovery studies by running spiked samples. These will usually show interferences, but may not detect the type of systematic bias which occurs when a sample component is tightly bound within a matrix and is not atomized efficiently. A spike of standard put into the same sample may be easily and efficiently recovered, because it is not bound in the same way as the sample component. The use of appropriate standard reference materials to test the method will be more likely to detect this kind of bias.
- Interferences in Atomic Absorption
While the method is generally quite specific, interferences can occur in atomic absorption spectrometry, both in flame and graphite furnace work. These may be classified as spectral, chemical, and ionization interferences.
- Spectral Interference:
Spectral interference arises when the absorbance (or emission) of an interfering species either overlaps or is so close that of the analyte that they cannot be resolved by the monochromator. Since the emission line from the hollow cathode lamp is so narrow, spectral interferences are not usually a problem. Two lines have to be less than 0.1 nm apart for this type of interference to occur. Spectral interferences can be readily eliminated by choosing a line further removed from the interfering line. For example, the vanadium line at 308.211 nm interferes with the aluminum line at 3082.15. This interference is easily avoided by measuring aluminum at 309.27 nm instead.
- Chemical Interference:
Species being measured may form refractory oxides which do not readily decompose into atoms. These require a higher temperature or a flame low in oxygen, so that oxide formation is reduced. For example, the formation of Ca(OH)2 would interfere in the analysis of other metals. This problem can be eliminated by substituting nitrous oxide for air as an oxidant. The higher temperature decomposes the Ca(OH)2 and eliminates the interference.
Chemical interferences occur when chemical processes during atomization change the absorbance characteristics of the analyte. One of the most common types of chemical interference is the formation of compounds of low volatility. For example, in presence of sulfate or phosphate, the calcium absorbance can fall significantly due to the formation of sulfate or phosphate species which will not vaporize at the flame temperature. In presence of aluminum, the absorbance of magnesium drops because complex oxides containing aluminum and magnesium form.
A releasing agent, for example, strontium or lanthanum, can be added to minimize the interference of phosphate in the determination of calcium. Here, the lanthanum or strontium ties up the phosphate and prevents it from combining with the calcium. A protective agent such as APDC (the ammonium salt of 1-pyrrolidinecarbodithioic acid) forms a stable, volatile complex which decomposes in the flame and prevents the formation of refractory compounds.
- Ionization Interference:
The high temperature in the flame can cause ionization of atoms. Thus the concentration of free atoms, which are the species being measured, decreases resulting in a lower sensitivity. The metal atoms are in equilibrium in the flame.
M Û
M+ + e- ( .19)
The equilibrium constant, K, for this reaction is given by:
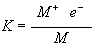 ( .20)
( .20)
From the above equation it can be seen that the metal atom concentration can be increased by increasing the concentration of free electrons in the flame. This is achieved by adding a ionization suppresser such as lithium. Lithium ionizes easily in the flame, supplying a relatively large quantity of free electrons. The ionization equilibrium shifts, increasing the concentration of free atoms.
- Background Correction in Atomic Absorption Spectrometry
If molecular species are formed in flames or in the cloud of atoms issuing from a graphite furnace, these will exhibit broad band absorbances rather than the line absorbances found with atoms. The bands may contribute substantial interferences across a fairly wide area of the available spectrum. Absorbance due to molecules formed in the flame without any contribution from the sample can be zeroed out as the blank is being aspirated. However, if materials contained in the sample interact to form interfering molecules, a background correction technique must be applied. The absorbance on either side of the line of interest can be measured, and subtracted as a background. This, of course requires a light source which emits at the needed wavelengths, so a continuous source such as a deuterium lamp must be used in addition to the line source lamp. Figure .26 shows the apparatus for this background correction method. A chopper alternately focuses the beam from the hollow cathode lamp and that from the continuous source through the sample and on to the detector. The radiation from the deuterium lamp is absorbed by the broad band interferents, and is measured and subtracted.
Another background correction involves the use of the Zeeman effect. When an absorbing or emitting atom is subjected to a strong magnetic field the line of radiation being emitted or absorbed is split into several lines. There are separated by only a few thousandths of a nanometer in wavelength. These extra lines arise from a change in the magnetic quantum number of an atom, in addition to the usual changes in electronic energy level caused by the absorption or emission of a photon. These additional side absorbance bands will absorb radiation polarized perpendicular to the magnetic field. The line arising from absorbance with no change in magnetic quantum number absorbs light which is polarized parallel to the field. The molecules responsible for the interference absorb the polarized light equally, regardless of its direction of polarization. This effect may be used to correct for background absorption by subjecting either the emitting atoms that form the light source or the sample atoms to a magnetic field. The Zeeman effect, then, gives a way of determining which part of the absorbance is due to the analyte atoms and which part is unaffected by the field. Therefore, part of the absorbance can be attributed to interfering molecules and subtracted as background.
Another background correction, the Smith-Hieftje method, uses the self absorption phenomenon which occurs when the lamp current is raised. This reduces the available light at the center of the band, while increasing it on either side of the spectral line. As the current to the lamp is increased, the background absorbance on both sides of the line is measured. Then the current is returned to the normal setting, giving the analytical absorbance. The background absorbance can then be subtracted to obtain the corrected absorbance. Zeeman effect, Smith-Hieftje, and deuterium background correction are available on commercial instruments.
- 3.8 Inductively Coupled Plasma Emission Spectroscopy
In atomic emission spectroscopy, a sample is often excited by exposing it to high temperatures. With solid samples an electrical arc or spark is used, but the spectrum obtained is difficult to use for quantitative work because it only lasts a second or so. However, if the sample is dissolved, the liquid solution can be aspirated into an argon plasma torch at 6000 - 8000oK. Because of the high temperature, the sample is very efficiently atomized, and the atoms are excited to an electronic level where they emit. Few molecular species are stable enough to survive this temperature and so interference from molecular emission bands is minimized.
To form the plasma, argon is passed at a high flow of 10-20 l/min through the torch. Surrounding the top of the torch tube is a water-cooled induction coil, powered by a radio-frequency generator and producing about 2 kW of energy, at around 27 MHz frequency. Ions formed by a spark passed through the argon are agitated by this strongly fluctuating field, and are forced to flow rapidly in a circular path. Friction heats the gas to the point at which it forms a plasma, which is a conducting gaseous mixture containing electrons and cations as well as undissociated atoms. Argon ions are stable enough that they will continue to absorb energy from the coil and stay hot enough to sustain the plasma indefinitely. The argon flow is directed through three concentric quartz tubes. The outermost one serves to cool the torch body as it would easily melt at the plasma temperature. The argon flowing through the inner tube is used to aspirate the liquid sample into the plasma. The plasma torch is shown in Figure .27.
The central core of the plasma, which appears as a bright white flame, emits a strong argon spectrum. About 20 mm above the core, the argon spectrum is much less strong, and the plasma appears transparent. This area is quite free of background lines and is suitable for analysis. The droplets of sample are exposed to the plasma for a couple of milliseconds before they reach this area, by which time they have been dried, evaporated, and atomized.
Since the torch provides a much higher temperature than an atomic absorption flame, chemical interferences are less of a problem. Also, ionization is not as serious a problem as one might expect, since the plasma contains a high concentration of free electrons. This produces a high enough concentration of excited atoms to give a sufficiently intense emission spectrum. Oxides do not readily form, as the plasma is relatively free of any active species, including oxygen. Therefore, the spectra emitted from the sample elements are quite clean and free of most interferences. Calibration curves tend to be linear over quite wide ranges.
The detectors and monochromators suited to atomic absorption spectroscopy are also used for ICP. Instruments for ICP are often combined with atomic absorption. This enables the use of the same monochromator and detector for both. Then a movable mirror either brings the beam from the hollow cathode lamp into the monochromator, or is switched to capture the emission from the plasma torch instead. The emitted lines are scanned as the sample is aspirated, or a diode array detector reads the lines simultaneously. Computer automated data collection on the ICP allows the comparison of emission at more than one wavelength for each targeted element, so that interferences can be detected.
Unlike atomic absorbance methods, ICP is an excellent tool for the simultaneous determination of multiple elements. The sensitivity is high, because the plasma gives a high atomization efficiency. In addition, molecular band spectra are much less of a problem because of the very high temperatures. Figure .28 shows an ICP spectrum of a sample containing copper, zinc and chromium.
Sample preparation is the same as that required for flame atomic absorption, except that modifiers for masking interferences or buffering ionization are not often necessary. The applications are also similar. ICP analysis is used for determination of most metals in water, soil and other environmental sample extracts or digests. The plasma torch is also used in conjunction with mass spectrometry. The ions formed in the plasma can be sent directly into the source of the mass spectrometer for detection by mass/charge ratio, instead of measuring the concentration by the radiation emitted. This technique is discussed in the mass spectrometry chapter.
- Comparison of Atomic Spectroscopic Methods
Table .5 shows the detection limits for some elements in the various atomic spectroscopy methods. It should be remembered, however, that the true detection limits can be influenced by the other components of the sample, which may interfere to some degree, and by the cleanness of the blanks, which can be the most difficult part of the analysis when ultratrace analyses are being done.
- 3.9 X-ray Fluorescence
When atoms are bombarded by x-rays interior electrons may be ejected. The remaining electrons cascade down to fill the vacancy. These electron transitions emit other x-rays with energies or wavelengths specific to the emitting elements. This phenomenon is known as x-ray fluorescence (XRF). It is especially suited to analysis of solids for their elemental composition. For screening for the presence of metals in reasonably high concentrations, for example for determining if lead is present in a painted wall, it is unsurpassed for ease of use, and is totally nondestructive. The great advantage of being able to analyze a solid directly without dissolving or extracting it, is counterbalanced by the difficulties of obtaining quantitative data from XRF and by its relatively low sensitivity. It is usually used for determining elements which are present in the 0.01 % range or above.
- Wavelength Dispersive XRF vs. Energy Dispersive XRF
In the x-ray region the grating is replaced by a crystal in which the planes of atoms serve to diffract the beam into monochromatic rays. Because of their much closer spacing, on the order of angstroms, these atomic planes can diffract radiation of very short wavelengths, such as x-rays. Some instruments are termed wavelength dispersive, and these contain a crystal which acts as a diffraction device to separate the various wavelengths of emitted x-rays.
However, since a photon in this region carries a relatively high amount of energy, it is possible to determine the energy of each photon as it strikes the detector. Therefore, one does not necessarily have to separate the rays of different wavelength in space, and scan them. All the emitted radiation can be directed to the detector, where the energy of each photon is determined as it impinges on the detector. These instruments tend to be simpler and less expensive than the wavelength dispersive x-ray spectrometers, but also provide lower resolution between closely spaced emission lines.
- X-ray Instrumentation
-
Sources
X-rays are generated by directing a beam of electrons at energies of 10-100 keV onto a metal target. The x-rays have a wavelength profile which depends on the target metal. A continuum of radiation is produced, with photons of discrete energies characteristic of the target superimposed on it. Higher atomic number targets give more intense radiation and the spectrum extends to shorter wavelengths as the energies of the impinging electrons are increased. It is also important that the target has a high melting point, so that it can stand up to the electron bombardment.
The selection of a target material can change the emitted spectrum, since the continuum wavelenths of the excitation are not as intense as the characteristic emissions. If the intense characteristic line is near an absorption band in the sample, the fluorescence will be enhanced.
Another means of producing the rays to stimulate x-ray fluorescence is by use of radioactive sources. These can be limited to a specific band of radiation by passing the radiation through two filters, one to absorb the energies below the desired range, and the other above the range. The specific wavelength band pass is tuned to the element to be detected, while the radiation returning to the detector is likewise selected. The radiation produced by the radioactive source is several orders of magnitude weaker than that of an x-ray tube. Therefore, the source, the sample and the detector must be located close together. However, the radioactive source, which needs no power, is ideally suited for hand held, portable screening instruments, tuned to a single element, often lead. The radioactive ring source with sample and detector is diagrammed in Figure .29.
- X-ray Detectors
Geiger counters, proportional counters and scintillation counters are used to detect the presence and intensity of radiation in the x-ray region of the spectrum. Geiger counters are simple to operate and do not require very sophisticated electronics. They have a rather slow counting rate and a relatively long "dead time", losing much of the radiation, even when the intensity is moderate. This dead time is the time between counts, when any radiation falling on the detector is not counted. Also, the Geiger counter is not capable of determining the energy of the measured radiation.
Proportional counters are useful over a similar spectral area as the Geiger counters, but have several advantages. The dead time is short, so the detector is suitable for use at higher intensities. The output voltage is proportional to the energy of the input x-ray photon, so energy discrimination is possible without a dispersing device.
The scintillation detector has very good sensitivity for x-rays with wavelengths below 2Å, and a very short dead time. It is capable of a rapid counting rate. The output voltage is proportional to the input ray's energy, so it can be used to reject radiation of energy bands outside that being measured. Its energy resolution is not as good as that of the proportional counter, but is useful for rejection of higher order bands of x-rays.
Many modern x-ray fluorescence spectrometers use semiconductor detectors. These are composed of silicon wafers, germanium crystals, or germanium crystals doped with lithium. The detectors are solids, in which electron-hole pairs are formed when the detectors are exposed to ionizing radiation. The electrons in the material are excited into the conduction band of the semiconductor by the absorbed radiation, where they move freely toward the positive electrode, producing a current pulse. The vacated hole travels in the opposite direction by repeatedly exchanging electrons with neighboring sites. Lithium-drifted germanium detectors require cooling with liquid nitrogen at all times after manufacture. The semiconductor is inherently unstable at higher temperatures and must be replaced if it is allowed to warm. Silicon based detectors are only useful down to wavelengths of 0.3Å. Germanium-based detectors are necessary at shorter wavelengths. Pure germanium detectors require cooling only during use, to reduce thermal noise, but not constant cooling, and so these are somewhat more convenient and less expensive to use.
These detectors have much greater energy resolution than the earlier types. For each photon absorbed a large number of electron-hole pairs are formed. The intensity of the pulse of energy formed in the detector is used to distinguish the wavelengths or energies of the radiation emitted by the samples. The resolution of semiconductor detectors enables discrimination between elements only one or two atomic numbers apart, making them useful for use in simpler, less expensive, qualitative and semi-quantitative analyzers. The basic layout of the x-ray fluorescence apparatus is shown in Figure .30.
- X-ray Fluorescence Samples
Sample preparation for x-ray fluorescence analysis is often very simple. Because the samples can be analyzed in the solid or liquid states, there is often no preparation at all. Samples such as airborne particulate can be captured on a filter and be inserted directly into the instrument for analysis. Liquids may be poured into holders with a thin mylar film forming the bottom. The exposure is then done through this window. Since several peaks may be produced for each element, these can be used to confirm identities of analytes.
While these simple methods are adequate for qualitative and perhaps semi-quantitative estimations, quantitative work must include consideration of the serious matrix effects which occur in x-ray fluorescence. Because the radiation released from the sample, the fluorescence, is absorbable by other elements in the sample, variations in the matrix will cause differences in the amount of interelement interferences.
First of all, the x-rays, especially at longer wavelengths, do not penetrate deep into most samples. Therefore the surface must be prepared and be representative of the whole sample. For most environmental samples, the sample will be a particulate material. Particle size and particle size distribution have substantial effects on the results of the analysis. To avoid matrix effects, samples may be diluted in a solid material such as sodium tetraborate or borax. The sample may be mixed with the diluent and formed into a pellet in a high pressure press. Alternatively, the sodium tetraborate may be melted with the sample and the fused mass used as the sample. The latter technique gives somewhat better results than pellet formation, because the fused sample is more homogeneous.
When trace materials are being sought, some concentrating technique is usually required. Airborne particles on a filter are used directly. The background due to the filter may be substantial, and filters of paper or organic membranes are generally more suitable than quartz and glass fibers. Ion exchange membranes may be used to collect ionic species from water samples. When the ions to be determined are a small part of the total ionic loading, as in sea water, chelating resins may be more suitable for collection and concentration of the sample. Interelement interferences can be reduced by keeping the sample thin, so that the excited emission does not have to pass through any substantial amount of the matrix before it is detected. If this is not possible, then the matrix should be comparable between the sample and the standard. Standards can be prepared for filters by drying known amounts of standard solutions on a filter, or by dispersing a known amount of the element in powder form on the surface of the filter. Calibration plots are often not linear and so calibration may have to be done at several different concentrations to determine the curvature of the plot.
If matrix interferences are minimized, and the sample and standard are similar in composition, a simple ratio of intensities of the emission for the sample and standard is adequate for quantitation. Internal standards, standard addition, and comparison with standards made up in a very similar matrix are all techniques that have met with success in x-ray spectroscopy. However, each of these must be looked at with care before use. Analysis of standard reference materials similar to the sample under consideration is a good way of determining whether or not there are substantial matrix or calibration problems. Mathematical methods are also developed which assist in the calibration and correction for interferences.
- 3.10 Hyphenated Spectroscopic Methods
The ability to determine both qualitative and quantitative information about a sample by examining its interactions with radiation make the combination of spectroscopy with separation techniques particularly valuable. Liquid chromatography uses both absorbance and fluorescence methods in detection, while infrared detectors have become useful in gas chromatography with the advent of Fourier transform methods. The complete IR spectrum of a peak can be obtained as it exits the GC column and a match with a computerized library can be done. GC/FTIR is almost as useful as GC/MS for environmental analysis, although it is inherently less sensitive. Since the IR is a nondestructive detector, it is possible to use both FTIR and MS in series after the gas chromatographic column. This gives the power to characterize most compounds by their mass spectra, to distinguish between isomers with similar mass spectra and to identify compounds by functional groups in the chromatogram, by IR.
- References and Suggested Readings
and
( .1)