Chapter
4
Chromatographic Methods
Chromatography, a group of methods for separating very small quantities of complex mixtures, with very high resolution, is one of the most important techniques in environmental analysis. The ability of the modern analytical chemist to detect specific compounds at ng/g or lower levels in such complex matrices as natural waters or animal tissues is due in large part to the development of chromatographic methods.
The science of chromatography began early in the twentieth century, with the Russian botanist Mikhail Tswett, who used a column packed with calcium carbonate to separate plant pigments. The method was developed rapidly in the years after World War II, and began to be applied to environmental problems with the invention of the electron capture detector (ECD) in 1960 by James Lovelock. This detector, with its specificity and very high sensitivity toward halogenated organic compounds, was just what was needed to determine traces of pesticides in soils, food and water and halocarbon gases in the atmosphere. This happened at exactly the time when the effect of anthropogenic chemicals on many environmental systems was becoming an issue of public concern. Within a year, it was being applied to pesticide analysis. The pernicious effects of long lived, bioaccumulating pesticides, such as DDT, would have been very difficult to detect without the use of the ECD. The effect of this information on public policy has been far-reaching.
The basis of all types of chromatography is the partition of the sample compounds between a stationary phase and a mobile phase which flows over or through the stationary phase. Different combinations of gaseous or liquid phases give rise to the types of chromatography used in analysis, namely gas chromatography (GC), liquid chromatography (LC), thin layer chromatography (TLC), and supercritical fluid chromatography (SFC).
Chromatography has increased the utility of several types of spectroscopy, by delivering separate components of a complex sample, one at a time, to the spectrometer. This combination of the separating power of chromatography with the identification and quantitation of spectroscopy has been most important in environmental analysis. It has enabled analysts to cope with tremendously complex and extremely dilute samples.
4.1 Principles of Chromatography
All chromatographic systems rely on the fact that a substance placed in contact with two immiscible phases, one moving and one stationary, will equilibrate between them. A reproducible fraction will partition into each phase, depending on the relative affinity of the substance for each phase. A substance which has affinity for the moving or mobile phase will be moved rapidly through the system. A material which has a stronger affinity for the stationary phase, on the other hand, will spend more time immobilized in that phase, and will take a longer time to pass through the system. Therefore, it will be separated from the first substance. By definition, chromatography is a separation technique in which a sample is equilibrated between a mobile and a stationary phase.
Gas chromatography employs an inert gas as the mobile phase, and either a solid adsorbent or a nonvolatile liquid coated on a solid support as the stationary phase. The solid or coated support is packed into a tube, with the gas flowing through it. Separation depends on the relative partial pressures of the sample components above the stationary phase. Liquid chromatography uses similar packed tubular columns and usually a pump to force a liquid mobile phase through the column. Supercritical fluid chromatography occupies a middle ground between gas and liquid chromatography. The mobile phase is a supercritical fluid, i.e., a fluid above its critical temperature and pressure. This allows the use of GC type detectors, since the mobile phase has gas-like properties, but also allows continuous variation in such mobile phase properties as viscosity and density, by changing temperature and pressure. Finally, chromatography may be done on a planar surface. The sample is transported over a solid surface such as cellulose or silica gel, coated on a plate. The sample components are moved over the surface by the mobile phase which is usually allowed to travel through the adsorbent layer by capillary action.
The reason that all molecules of a certain type tend to exit the system at the same time is that they are always re-equilibrating between the phases. Over a large number of such equilibrations, the molecules spend, on average, the same amount of time in each phase. Let us look at one point in the chromatographic column. When the analyte achieves or approaches equilibrium, the mobile phase moves on, leaving the stationary phase with too much of the analyte and the mobile phase with too little. To attempt to reestablish equilibrium, more sample dissolves in the mobile phase and moves along. Figure Chapter 4 .1 shows a mixture of three substances as they move through a chromatographic column.
The movement of analytes in the column can be described mathematically. The basis of chromatography is the equilibrium of each analyte between the mobile and stationary phase. This can be expressed by a simple equilibrium equation, where Kx is partition coefficient,
Kx = [C]s / [C]m ( Chapter 4 .1)
that is: the concentration in the stationary phase ([Cs]) is directly related to the concentration in the mobile phase ([Cm]), at least when the concentrations are low. Chromatographic separations are best done with a small amount of analyte, which keeps either phase from becoming saturated with analyte, so that the concentrations in the two phases are directly proportional. Overloading the column with sample causes one of the phases to become saturated with sample, leading to a loss of column efficiency, and poorly shaped peak profiles.
The quantities in the equilibrium expression for Kx, [C]s and [C]m are not easy to measure. We can define a new constant, the capacity factor, k':
k'x = (moles of X in stationary phase)/(moles of X in mobile phase) ( Chapter 4 .2)
Since the number of moles can be expressed as the concentration multiplied by the volume, Equations 6.1 and 6.2 can be combined and reduced to:
k'x = Kx (Vs/Vm) ( Chapter 4 .3)
All sample molecules spend the same amount of time in the mobile phase. If they were completely unretained by the stationary phase they would exit the column in the time it takes for one volume of mobile phase to pass through the column. This is equal to the void volume of the column. Molecules pass through the column in the time equal to the passage of one void volume, Vm, plus the time spent in the stationary phase, expressed by k'x. Therefore, the volume of eluent which will pass through the column before the sample elutes (the retention volume, Vr) can be expressed as:
Vr = Vm ( 1 + k'x ) ( Chapter 4 .4)
The retention volume, Vr, is related to the column flow Fc, and the retention time, tr. Likewise, the volume of the mobile phase, Vm, is related to the flow and the time the void volume takes to pass through the column, to.
Vr = tr Fc
(
Chapter 4 .5)
Vm = to Fc ( Chapter 4 .6 )
Substituting these into equation 6.4 and rearranging gives:
k'x = (tr - to) / to ( Chapter 4 .7)
Values for all these variables can all be obtained from the experimental chromatogram, as shown in Figure Chapter 4 .2. The term (tr - to) is called the adjusted retention time and is often expressed as t’r, so k’x can also be expressed as t’r/ to. This is then the basis for separation of any two analytes. The separation is directly related to the difference in the k’ values for the two substances. If the k’ for the sample components is very small, there is so little retention of the compound that separation is not possible. If the difference between the k’ factors for two compounds is small, separation of them will be difficult. The selectivity, a, of a column for a particular separation, say of substances A and B, is expressed as a ratio of their retention times or retention factors:
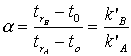 (
Chapter 4 .8)
(
Chapter 4 .8)
One should notice that the sample bands tend to spread out as they move through the column. The narrower the initial band of sample, and the less the individual compounds are spread out as they traverse the column, the more efficient the column is. An efficient column can separate a greater number of individual compounds in a given time.
Column Efficiency
By analogy to the process of distillation, the separating power of a column is expressed in terms of "theoretical plates". This term refers to the length of column in which the analyte equilibrates between the two phases. The more efficient the column is, the smaller the height of the plate will be, and the more equilibrations will occur in the length of the column. An increase in efficiency is most easily seen as a decrease in the width of each sample peak, showing that the bands of sample have not spread much as they passed through the column. A chromatography peak can be approximated to be a normal or Gaussian peak, and “height equivalent of a theoretical plate” H is defined as the variance per unit length. This is a measure of band spreading per unit length of the column. The separation capability of a column is expressed as the number of “theoretical plates” or N, i.e., number of plates in a column. The number of plates contained in the column, is made evident in the peak width of the sample components. Therefore, the number of plates, N, and H can be calculated from a test chromatogram.
N = 16 (tr/w)2 or N = 5.54(tr/w½)2 ( Chapter 4 .9)
where w½ is the peak width at half height, and
H = L/N ( Chapter 4 .10)
where L is the column length.
The efficiency of a column is a function of several parameters. These include the size of the column packing particles, the uniformity of the packing, the flow of eluent, and the rapidity with which equilibrium is established between the two phases.
Two molecules, moving through the column in the eluent flow, may find different paths, especially if there are gaps in the packing which allow eddies or swirls in the eluent flow. Molecules caught in these eddies will be slowed in their movement through the column, and will therefore elute on the back tail of the compound peak. If molecules can take a variety of paths of different lengths through the column, the peak will be broadened. Figure Chapter 4 .3 shows the phenomenon of eddy diffusion.
Band broadening is also due to the fact that a solute in a gas or liquid stream has a natural and unavoidable tendency to diffuse both forward and backward in the stream. The only factor which can change this is the viscosity of the mobile phase, which is not usually very easy to change. A faster flow, however, gives the peak less time to diffuse.
The mass transfer kinetics between the two phases also have an effect on the band width. Equilibrium between the mobile and stationary phases is never quite achieved in a chromatographic system. The further from equilibrium the system is operating, the poorer the efficiency will be. Diffusion of the sample band in the mobile phase and in the stationary phase both have an effect. Solute molecules may be held up excessively when they become trapped in deep pools of stationary phase, or in stagnant portions of mobile phase. If a substantial fraction of the molecules encounters such delays, the result is a spread-out peak. Figure Chapter 4 .4 shows cross sections of support particles coated with a liquid stationary phase, and how molecules may be excessively delayed. These pools and backwaters may be avoided by spreading a very thin film of support on a fairly regular shaped particle. Of course, this thin phase has little volume, which makes k' smaller, and leads to easy overloading of the column. Figure Chapter 4 .5 shows how the maximum concentration of the solute in the mobile phase is slightly ahead of that in the stationary phase. The bigger the difference in the location of these peaks, the wider the final exiting sample band will be. If the flow is too fast for the equilibration rate or vice versa, it will cause band broadening due to mass transfer effects.
The plate height, H, is a function of the flow rate of the mobile phase expressed as linear velocity, v, and can be calculated from the equation:
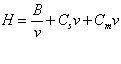 (
Chapter 4 .11)
(
Chapter 4 .11)
The B term describes the longitudinal diffusion, and is related to diffusion of the analyte in the mobile phase. This term becomes smaller as the velocity increases, because less time is available for the solute to diffuse. The Cs term is related to mass transfer in the stationary phase, and Cm to mass transfer in the mobile phase. Both of these terms increase with flow, because the solute is less able to reach equilibrium between the two phases as the flow becomes faster. The contribution to the overall H of the column by each of these factors can be plotted against flow velocity. Figure Chapter 4 .6 shows such a plot. The H line shows the combined effect of the factors. The minimum in the curve indicates the flow velocity which will give the maximum efficiency in that particular column. Using a carrier gas flow which is higher than the optimum will cause some loss of efficiency. However, this may be a satisfactory compromise, since it will also reduce the retention times, and so may shorten the time required for an analysis. Using a flow slower than the optimum is, however, never a good idea, since it lengthens the analysis time and also causes a loss in efficiency.
Although information on the optimal flow rate is often supplied by the column manufacturer, the best flow can be determined experimentally. The value of H can be found at several different flows, and a plot constructed for the column.
If there is a difficult separation to be done, there are two approaches. The column material may be changed, which will change both k'A and k'B, in order to produce a larger difference between them. On the other hand, if a more efficient column is used, the same amount of difference in retention time will give a more complete separation, because each peak will be narrower. Figure Chapter 4 .7 shows a poor separation which is improved in one case by increasing the value of a, which moves the peaks apart, and in the second case, by improving the efficiency of the column, which makes the peaks narrower, while not making the retention time difference any greater. Increased efficiency can take the form of either lengthening the column, which has the drawback of also increasing analysis time, or decreasing H by improving the quality of the column itself.
The degree of separation between any two peaks, A and B, can be expressed as the resolution of the peaks. This is defined as the difference between the two retention times divided by their average peak width.
![]()
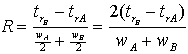 (
Chapter 4 .12)
(
Chapter 4 .12)
Alternatively, resolution can be expressed in terms of the selectivity factor and the capacity factors of the two substances being separated:
![]()
![]() (
Chapter 4 .13)
(
Chapter 4 .13)
where a and k’ are averages (since the values for these factors are similar, if the retention times are close, as a consequence of Equations 6.7 and 6.8).
From this equation the interaction of the three factors which influence resolution can be seen. A low value of k’ for the compounds being separated will lower resolution, since separation cannot take place efficiently if the analytes are not retained long enough to be separated. If a, the selectivity, is not sufficiently large, the compounds are only slightly different in their retention properties and are therefore very hard to separate. Finally, the resolution depends on N, the number of theoretical plates in the column, a measure of the efficiency of the column, indicating that a more efficient column can accomplish a separation even if a or k’ are lower than one would wish.
The same relationships can be expressed in terms of retention time rather than resolution. The retention time of the second of two peaks, A and B, is given by
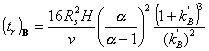 (
Chapter 4 .14)
(
Chapter 4 .14)
where v is the carrier gas velocity.
Example:
Heptane and toluene were
separated with retention times of 15.4 and 16.5 min respectively on a 1.0 meter
packed column. An unretained species passed through the column in 1.8 min. The
peak widths measured at the base were 1.15 for heptane and 1.20 min for
toluene.
a) Calculate the
resolution between the peaks:
![]()
b) Calculate the average
number of plates for the column.
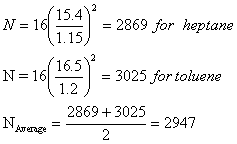
c) Calculate the average
plate height.
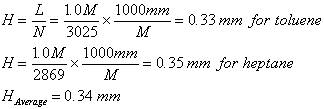
d) Calculate what column
length will be necessary to achieve a resolution of 1.5 on this column.
Since k’ and a do not change with column length, Equation 4.13
indicates that the resolution is proportional to ![]() . So,
. So, 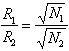 and
and ![]() . N2 =
7504.
. N2 =
7504.
Since L=NH, the length required to generate this number of plates is :
L =
7504 x 0.34 =
2551 mm or 2.5 meters
e) Calculate the capacity
factor for toluene:
![]()
The General Elution Problem
There is a problem which arises in all types of chromatography,
when samples of widely differing retention properties are present in the same
sample. If the elution conditions are correct for the early eluting compounds,
the late ones will remain in the column too long. They will be so broadened
that it will be difficult to determine their area accurately. Indeed, they may
not elute at all. On the other hand, if we set up the system so that the later
eluting compounds spend less time in the stationary phase, the early peaks will
come out so quickly that they will not have sufficient time in the stationary
phase to allow adequate separation. This problem is called the ‘general elution
problem’ and is solved in different ways in different types of chromatography.
Usually, some type of "programming" is done. This involves a gradual
or stepwise change in one of the operating parameters. The best conditions for
the separation of the early compounds are set up. Then these are changed during
the run to conditions better for the elution of more retained compounds. In gas
chromatography, the column temperature is raised over time until a temperature
favorable for the separation and elution of the later peaks is reached. This is
called temperature programming. In
liquid chromatography, where retention is more dependent upon strength of the
mobile phase, the composition of the mobile phase is changed as a function of
time. This is called gradient
programming.
4.2 Quantitation in Chromatography
The amount of each component in the sample is measured as it issues from the column, by passing the effluent through a detector and integrating the detector signal over time. For quantitation in both gas and liquid chromatography, the detector signal is usually fed into a digital or analog recorder. The strip chart recorder produces the typical chromatogram, a plot of signal versus time, as a series of peaks. The peak area is related to the quantity of each material, and the location on the time axis or the retention time is used to identify the compound. Modern chromatographs have high precision in retention time as well as peak area. Consequently, chromatograms provide excellent qualitative and quantitative information. Digital processing of the signal allows the peak areas to be integrated automatically, with the beginning and ending points of peaks indicated. Baselines are usually set automatically and may sometimes be adjusted by the operator. Peak areas obtained on strip chart recorders may be determined graphically by drawing tangent lines to the peak sides and measuring the widths and heights as was indicated in Figure 4.2. The area (A) is calculated from:
A = hmax x w½ or (hmax x wb)/2 ( Chapter 4 .15)
In addition, a peak area may be measured by cutting the peak out of the chart paper with sharp scissors, and weighing it on an analytical balance.
In order to calculate the quantity of sample from the peak area or peak height, the detector must be first calibrated by running standards. Quantitation may be done by either peak height or peak area, with most accurate results being achieved with use of areas, although, when chromatograms are being manually interpreted, heights may be more readily measured. External standards, internal standards or a combination of both can be used.
External Standard Method
A series of standards containing known quantities of the analytes are prepared and run. The peak heights or areas are plotted versus the quantities or concentrations, and the samples are run in the same way. A major source of error in this method is the reproducibility of the injection, especially if manual injections are being made using a syringe. Since most chromatographic samples are only a microliter or less, accurate measurement is difficult. Automatic injectors and sampling valves can reduce this error to a few percent.
The concentration of the sample is calculated from the calibration graph or from:
Cunk/Cstd = Aunk/Astd ( Chapter 4 .16)
where C is the concentration and A is the peak area for the standard (std) and the unknown(unk). This simple calculation is usually good enough if the sample and standard are of the same order of magnitude and the detector response is linear over the range of concentrations covered by the sample and standard. If sample concentrations vary over a wider range, a calibration curve should be constructed over the entire range.
Since it is not always possible to obtain standards over a suitable range or for every compound one needs, detector response factors may be used. These allow calibration with one compound as a surrogate for others.
Cunk / Cstd = funk Aunk / Astd ( Chapter 4 .17)
where funk is the detector response factor which relates the detector response to a quantity of the sample compound to its response to the same amount of the surrogate standard compound. These factors can be determined experimentally. Then the chromatograph can be calibrated daily against the surrogate compound, and the detector response factors used to calibrate for other compounds. It is best to use compounds for standards which resemble the target compounds as much as possible, being of similar molecular weight and polarity, to reduce possible errors.
The Internal Standard Method
An internal standard is added to the sample before analysis. This is composed of one or more compounds with sufficient similarity to the target analytes, so that they behave similarly, but must surely not appear in the samples. Internal standard compounds must also be readily separated from any of the compounds in the sample. If an internal standard is added before such steps as concentration, extraction, or dilution, it is then not as necessary to make accurate volume measurements, and the injection volume is also not as critical. By examination of the peak area of the internal standard in sequential injections, the reproducibility of the injection volumes can be seen. Small changes in the volume show up as changes in the internal standard peak. These can be compensated for in the calculations, by working with the ratio of the sample peak/internal standard peak, rather than just the peak areas. Again, it is important to keep the concentrations of the internal standard and the target compounds in a similar range, to keep linearity problems from arising. The area of the internal standard (Ais) is used to normalize the areas of all other sample peaks, thus eliminating the effect of differences in injection volumes or dilutions.
Cunk = ( Aunk / A is ) Cis funk ( Chapter 4 .18)
4.3 Gas Chromatography
In gas chromatography, the eluent is an inert gas, often helium, hydrogen or nitrogen. The eluent actually has little effect on the separation process, which is governed more by the volatility of each sample component and its interaction with the stationary phase. Stationary phases are either solids or liquids and are contained in columns which range in internal diameter from 100 micrometers to 4 mm. The chromatographic system consists of three essential elements: an injection system, a temperature controlled column, and a detector. To this basic system, many enhancements can be added. The column oven may be equipped to carry out temperature programming and cryogenic cooling Additional detectors may be added, and an automated injection system, computerized instrumental control and data analysis systems may also be installed. For a chromatograph to be used in environmental analysis, specialized injection systems, such as concentrators or thermal desorbers for air analysis, or purge and trap apparatus for water analyses are often useful. In addition, complex environmental samples often require a detector or array of detectors to assist in identification of the sample components as well as to determine their concentrations.
Injection Devices
An injector which can place a small, narrow, and reproducible band of sample on the head of the column is absolutely essential to good chromatography. The more efficient the column is, the more demand is placed on the injector to keep the initial sample band narrow. The sample band will only become wider as it traverses the column. If it is wide to begin with, the efficiency of the column will be overshadowed by the band broadening which takes place outside the column, and which contributes nothing to the separation. This is known as extra-column band broadening.
A syringe is the traditional injection device in GC. A heated injection port, equipped with a soft polymeric septum, is located at the head of the column. The sample is injected through the septum into the heated carrier gas stream, and vaporizes. The carrier gas flows through the injection port and sweeps the sample onto the column. The injection port must be well designed so that the sample is quickly and efficiently moved onto the column, with all interior areas rapidly swept out by the carrier gas. Unswept areas allow the sample to become trapped, and diffuse slowly back into the gas stream, causing peak tailing. A glass liner is often incorporated into the injection port, so that it may be removed and cleaned or replaced if samples leave a non-volatile residue.
Since a high efficiency column may require a sample of only a few nanograms, a syringe may not produce adequately reproducible samples. A splitting injector port may be used. In this case the sample is injected into a chamber where it is mixed with a volume of carrier gas. Then a small part of this mixture is allowed to flow into the column, and the rest is vented. An injection port and a splitter are shown in Figure Chapter 4 .8.
When the sample to be injected is a gas, a gas sampling valve is often used to inject a measured portion into the column. This valve contains a loop of known volume which is filled in the ‘load position’ by allowing the sample gas to flow through it. When the valve is turned to the ‘inject position’, the carrier gas is diverted through the loop, and the sample is carried onto the column. In Figure Chapter 4 .9, you can see that the carrier gas always flows to the column, whether the valve is in the load or inject position. The sample size is usually changed by installing a different sized loop, although, if a pressure gauge is mounted on the system, the sample pressure may be changed to change the amount injected. Since the sample is a gas, the temperature of the loop will also make a difference, if it is subject to wide variations.
More elaborate injection systems are found in instruments dedicated to a particular determination. A thermal desorber, for instance, is employed for transferring samples from an adsorbent trap into the column. A purge-and-trap apparatus is used for stripping volatile compounds from water or sludge samples, and injecting them into the column. These will be described in the chapters on air and water analysis.
Columns
There is a vast selection of GC columns available. Many of them are so slightly different from others that they are readily interchangeable. The columns differ in physical dimensions. Large diameter packed columns, having a larger volume of stationary phase, are able to accommodate large samples. The smallest columns, open tubular capillary columns, have the highest efficiency, but are limited to tiny samples.
Chromatographic stationary phases may be either solid or liquid. When a solid stationary phase is used, adsorption is the retention mechanism, and the technique is called gas solid chromatography. It is used mainly for low molecular weight volatile organic compounds and gases such as CO, CO2 or SO2 or H2S. The stationary phase may also be a viscous liquid coated or chemically bonded to the surface of a packing material, or on the inside wall of a open tubular capillary column. This is called gas liquid chromatography (GLC). The mechanism of retention in a liquid stationary phase involves partitioning of the analyte between the gas and the stationary phase. Liquid phases must have good solvent characteristics, should be inert and stable at relatively high temperatures.
Table Chapter 4 .1 shows a selection of liquid stationary phases, with the kinds of samples for which they are suited. As gas chromatography developed, hundreds of stationary phases were described in the literature. To characterize these in a quantitative fashion, Rohrschneider developed a series of constants to describe the selectivity of a column toward different types of sample. A suitable phase for a particular sample can be selected on a rational basis by using these constants. This work was extended and refined by McReynolds, and the McReynolds constants for stationary phases are listed in the catalogs of almost every supplier of chromatographic columns.
The McReynolds constants are based on the assumption that different molecular interactions, such as dispersion, orientation, induced dipole, and donor-acceptor complexation, between functional groups on the column material and those of the sample are additive. A series of probe compounds was chosen to represent each type of interaction, and the retention time for each probe is determined on the column in question. Since retention times change with phase loading, temperature, and flow, a retention index for each probe is determined by injecting the probe compound along with a set of normal hydrocarbons which will bracket the probe compound's retention time. The retention index is calculated from the following equation:
![]()
![]() (
Chapter 4 .19)
(
Chapter 4 .19)
where each t'R is an adjusted retention time. The unknown is indicated as x, and z is the number of carbons in the normal hydrocarbon eluting just before the sample. The hydrocarbon, z+1, is that which elutes just after the sample. This index is not entirely independent of column conditions, but is still useful. The McReynolds constant is expressed as the retention index of each probe compound on the tested column minus that on squalane. The probe compounds, and the interactions they indicate, are listed in Table Chapter 4 .2.
These constants are useful in comparing columns, and in selecting a new column to improve a particular separation. For instance, if alcohols are to be separated, one would look for a column which has a large Y constant, indicating good retention and selectivity for alcohols. When a column described in the literature for a particular analysis is not available, a suitable substitute can be chosen, by selecting one with similar McReynolds constants.
Packed Columns
Packed columns vary in internal diameter, with most being constructed from 1/4 or 1/8 inch OD tubing. They also vary in the particle size of the packing, and the amount of stationary phase coated on the support. One of the parameters usually listed is the percent loading. The loading is determined by the mass of stationary phase coated on a given mass of support. Loading ranges from a few percent to 20 or 30%.
Stationary phases are usually coated onto an inert support material, which should be of uniform size and good mechanical strength. The efficiency of the finished column depends on both the particle size and uniformity of the size. Smaller particles give more efficient columns, but if the particles become too small, the flow through the column at reasonable gas pressures is hindered. Usually column packings in the 60-80 mesh or 80-100 mesh ranges are used. These are often made from treated diatomaceous earth, a silica based material.
Supports vary in their surface activity and many treatments have been developed to modify the surface. The support may be extracted with acid to remove surface metal ions which may form active sites on the packing, adsorbing sample molecules too tightly and causing tailing. The support may also be treated with a silanizing agent to further reduce surface activity. For extremely polar compounds like water or sulfur dioxide, an extremely inert support is made from granulated fluorocarbon polymer.
The packing is covered with the stationary phase, generally by mixing the support with a solution of the stationary phase, a high boiling liquid, in a volatile solvent. As the solvent is evaporated the packing is coated with an even layer of stationary phase. When the solvent is dried off, the packing should be free-flowing and easy to pack into the column without gaps or loose spaces.
Stainless steel or glass tubing is used to form the columns. Stainless is considerably easier to handle, as it is not breakable and can be gently bent to fit the instrument at hand. Glass columns are usually deactivated by being silanized, and have a lower surface activity, so may be needed for especially sensitive samples, such as very labile organic compounds. It is important to handle packed columns fairly gently, and bend them carefully. Rough treatment will fracture the packing particles, increasing the non-uniformity of the packing by making smaller particles.
For gas-solid chromatography, several types of solid column packings are available. These may be porous polymeric materials, silica, or carbon based solids. Solid phases may be more durable, and able to stand up to some fairly aggressive samples, since they have no surface coating. They are usually used for small organic molecules and inorganic gases. Molecular sieves are used for gases such as CO, CO2, and O2.Some packings are designed for specific types of samples, such as water and alcohols, amines and nitro compounds, or sulfur gases.
Polymeric packing materials differ in their surface polarity and their pore sizes. They are especially useful for gaseous samples and for aqueous samples. Recently, carbon based materials have become quite popular. These are manufactured specifically for chromatographic separations and vary in such parameters as the pore size, the surface area per unit mass, and the particle size. Table Chapter 4 .3 lists some of the more common types of solid supports.
Open Tubular Columns
Open tubular columns are often used for complex mixtures, where large numbers of components must be separated. These are made of fused silica tubing, coated with a thin film of stationary phase on the interior, and covered with a polyimide coating on the outside surface. The polyimide is necessary to provide mechanical strength as fused silica can break easily if it is scratched. Recently, open tubular columns are also being made with silica lined stainless steel tubing. Choosing the best stationary phase is done in the same way as for packed columns. Selection of column diameter and phase thickness requires consideration of the sample to be separated. The narrowest diameter columns give the highest separating power, but the sample size is very limited. When trace components are to be determined, there may not be enough sample to detect. The thinnest coatings of stationary phases also give highest efficiency, but again limit the size of the sample which can be analyzed.
Many available capillary columns have chemically bonded phases. In these, a chemical reaction takes place between the liquid phase and the surface after the column is coated. The liquid forms chemical bonds to the hydroxyl groups found on the surface of the fused silica. These phases have several advantages. They are mechanically stable, and will not tend to creep towards the lower part of the column, even at high temperatures. This helps to extend the useful life of the column. If a bonded column becomes contaminated, which may happen especially easily when it is used for direct on-column injection, it may be washed out with solvent. This will often return the column to its original efficiency.
Internal diameters of capillary columns range from the highest efficiency ones of 0.15 mm to wide bore columns of 0.53 mm. Typical values for H range from 0.13 mm for a 0.15mm ID column to 0.45 mm for a 0.53 mm ID column. The column length selected again depends on the complexity of the sample and the number of plates needed to effect the separation. The shortest column which will accomplish the separation is best, since a longer column will require a longer analysis time without adding to the information gained. One virtue of capillary columns is the ease of dividing a long column into two shorter ones. Sometimes a long column, which has been shown to have an excess of theoretical plates for the use, may be cut in half.
The usual liquid stationary phase layers are in the range of 0.12um to 0.2 µm. The thickest stationary phase coatings may range up to 5 mm, and find their most usual usage in the analysis of very low boiling substances, especially samples which are gases at room temperature. Figure Chapter 4 .10 shows the effect of film thickness on separation, and Figure Chapter 4 .11 shows the effect of column diameter. The best separation is that which gives an adequate separation in the shortest time.
Porous layer open tubular (PLOT) columns are somewhat of a hybrid between packed and capillary columns. These are open tubular columns with a solid stationary phase layered on the inside surface of the column. These provide a means of doing gas/solid chromatography, with the advantages of an open tubular column.
The considerations which go into the selection of a column for any particular analysis can be summarized as follows:
· Is there a column offered which is designed for this analysis? Suppliers often design and sell columns which have been optimized for a certain commonly done sample. For example columns are made for light hydrocarbons, gasoline, pesticides, fatty acid methyl esters, and amines.
· Is there a column specified in a reference in the literature for a similar analysis? If so, the same column or one with similar McReynolds numbers should be satisfactory.
· A rule of thumb for column selection relies on the fact that the separation takes place in the stationary phase. Therefore, a phase which has a higher affinity for the sample components will be better for the separation. Relying on the maxim "Like dissolves like", a nonpolar column will do best for nonpolar samples and polar samples should be separated on a polar phase.
· Column length, diameter, and film thickness depend on the complexity of the sample, as well as the amount of sample which will be injected and the boiling range of the sample.
Column Temperature
A column has a minimum and maximum use temperature, which depends mostly upon the stationary phase. The minimum temperature is usually the freezing point of the stationary phase, since the solid has quite different properties from the liquid, and does not always partition the sample adequately. The maximum temperature is usually that at which the column bleed becomes intolerable. This is usually due to thermal breakdown of the stationary phase, or to the boiling off of the lower molecular weight fraction of the phase.
The temperature for the analysis must be optimized in the development of the method. The usual method is an educated trial and error approach. Equations exist which attempt to predict retention times based on column temperatures and thermodynamic properties of the sample components, but, in practice, these are seldom used. Generally, a standard or sample containing the components of interest is run on the selected column, at a temperature selected on the basis of the range of boiling points of the sample. It is not necessary to bring the column up to the boiling point of the higher boiling components. Compounds will migrate through the column at temperatures much lower than their boiling point. Thinner phase columns will require lower temperatures to elute peaks than those with thicker films. Referring to Figure 4.10, we can see that the 0.4 mm film column eluted the test peaks much more rapidly than the 1.2 mm film column. If the temperature on column the 1.2 mm column were raised, the chromatogram would begin to appear more like that produced on the 0.4 mm column.
When the last peaks in a chromatogram elute late and are very broad, the problem may be corrected by raising the column temperature. However, this often causes the earliest peaks to be unresolved because they are not retained long enough. In this case, a temperature program must be used. The sample is injected with the temperature optimized for the separation of the earliest peaks. The temperature may be held until the first peaks are eluted, and then the temperature is ramped up to a temperature which will bring out the last peaks, separated, but not excessively broadened. The rate of temperature rise depends on the number of peaks eluting during the ramp. If few peaks are present, then a rapid ramp might be chosen, while a complex sample with many components, requires a slower ramp rate. Figure Chapter 4 .12 shows a standard containing four hydrocarbons. In the isothermal run, the early peaks are sharp and well resolved but the late ones are broad. When a temperature program is used, a better chromatogram is obtained, and the time to resolve the four compounds is halved.
Generally, a few test runs will be needed to determine the best temperature program. Some chromatographic instruments are capable of multiple ramps and pauses during a program. These can be used to fine tune specific parts of a separation. For example, let us suppose that both the early and late parts of a sample are well separated, but there is a pair of peaks in the middle which are overlapping. One might slow the rate of temperature rise before these peaks elute, to retard them a little longer in hopes of improving the separation. After they elute, the oven temperature is raised to the same final temperature as in the original program.
4.4 GC Detectors
The eluent from the column is directed to one or more detectors. These produce signals which are proportional to either the amount of sample present in the detector at any moment, or the concentration of sample in the detector. The detector signal is usually displayed as a plot of signal magnitude versus time, giving the classic chromatogram. Detectors vary in their response to different classes of compound, from the thermal conductivity detector, which is universally responsive to all compounds, to such specialized detectors as the flame photometric detector, which detects only sulfur or phosphorous containing compounds, depending on the way it is set up. The selectivity of a detector is usually expressed by the ratio of the response to the desired analyte divided by that towards an interfering compound. For instance, the selectivity of a sulfur specific detector may be given by its response to a nanogram of sulfur divided by that for a nanogram of hydrocarbon. Since the interfering material may be present in much greater quantity than the analyte, selectivity factors of 104 or more are desirable.
The characteristics sought in a gas chromatographic detector are high sensitivity, a linear dynamic range of 4 orders of magnitude or more, a favorable signal to noise ratio, and good long term stability. A small detector dead volume is also important. If the sample has an opportunity to mix with a volume of carrier gas before the detection process is completed, the peak will be broadened and efficiency lost.
Thermal Conductivity Detector (TCD)
The TCD is a truly universal detector. It consists of a heated sensor in a thermostated chamber, through which the effluent flows. Helium is usually used as a carrier gas, as it has the highest thermal conductivity of any gas, except for hydrogen. As the peaks elute, the thermal conductivity of the gas in the chamber changes. This changes the heat flow from the heated sensor, through the gas, to the walls. Since the sensor is being heated at a constant rate, it becomes hotter as the thermal conductivity of the effluent drops. The change in temperature of the sensing wire filament or thermistor changes its resistance. The sensor is wired into a Wheatstone bridge circuit, and the change in resistance produces an unbalance, which produces a signal. The circuit for the TCD detector is diagrammed in Figure Chapter 4 .13.
The filament is sensitive to oxidation while heated, and therefore must not have current flowing unless the carrier gas is passing through the chamber. The detector is limited by its relatively low sensitivity, compared to other detectors, and usually has a fairly large dead volume. It is, therefore, not very suitable for capillary work. Because of these limitations the TCD is little used in environmental work, except for the determination of major constituents of air.
Flame Ionization Detector (FID)
The FID is a major workhorse of environmental analysis. It is nearly universally sensitive to organic compounds, and shows good sensitivity and excellent linearity. The column effluent is fed into a flame fueled by hydrogen, with a forced air flow. Figure Chapter 4 .14 shows a typical FID. A potential of several hundred volts is imposed between the tip of the flame burner and the collector which surrounds the flame. As the sample components burn, they produce a burst of ions. These produce a tiny current between the flame tip and the collector. The current is amplified by a high impedance electrometer and measured. The background current flowing in the detector is in the region of 2 x 10-14 to 10-13amp. In the presence of organic compounds the current will rise to 10-12 to 10-9amp. The response of the detector will change if the flows of air and hydrogen to the flame change. These flows should be checked for consistency when the detector is calibrated and used.
The FID detector has a number of advantages. The response is roughly proportional to the number of carbon atoms in the flame at any time, although certain substituent atoms, such as chlorine, reduce the response. The detector is insensitive to inorganic gases, water, carbon dioxide, sulfur dioxide, nitrogen oxides and other non-combustible gases. The detector has a very wide linear range, over about 7 orders of magnitude, has a low dead volume of about 1 ml, and is relatively noise free and easy to operate. Its major disadvantage is that it destroys the sample, so it cannot be passed on to another detector. Also, the fact that it requires both compressed air and hydrogen, as well as carrier gas, can be an inconvenience, especially when instrumental portability is an issue.
Electron Capture Detector (ECD)
This is probably the most used of the compound class specific detectors for environmental analyses. It has been used to trace the fate of such pesticides as DDT, as well as the halocarbon gases in the atmosphere. Its response depends on the electron-capturing properties of the sample. The detector is highly sensitive to the presence of electron capturing substituents, such as halogens, peroxides, and nitro- groups, on the sample molecules. When one looks at lists of regulated compounds, it is striking that so many of these are electron capture active.
The detector (Figure Chapter 4 .15) consists of a chamber containing a ß-emitting foil, usually nickel-65. This radioactive source emits electrons which ionize the carrier gas, and form a small current between the electrodes in the chamber. When electron- capturing species are present, they reduce this current. The reduction is detected by the electronics and measured.
The detector response varies widely, depending on the electronegativity of the species being detected. Table Chapter 4 .4 shows the relative sensitivity of the detector towards a variety of substances. We can see that the response to the individual electronegative groups depends upon their number in a molecule as well as their location. The response to chlorinated hydrocarbons increases by about a factor of ten with each additional chlorine atom present in the molecule, for example.
If the carrier gas forms metastable ions, it may cause undesirable collision reactions. Therefore, helium cannot be used with the ECD detector. Nitrogen is suitable, and a mixture of argon with 5 - 10% methane is also sometimes used. If a capillary column is being used, extra gas, called make-up gas must be added to the detector to bring the flow up to that for which the detector is designed. This helps to minimize band broadening. In this case, if nitrogen is used for the make-up gas, helium may be used for the column carrier gas.
Since ECD's contain a radioactive isotope, they are subject to governmental regulation. Depending on the design of the unit, a license may be necessary before one can be purchased. There are tests, called wipe tests, for leakage of radioactivity which must be done at specified intervals, to comply with regulations. The company which sells the unit will supply information on required licensing and routine testing.
The sensitivity of the ECD toward halogenated hydrocarbons and many pesticides is extremely high. The response to a particular compound is usually quite temperature sensitive, and the detector's linearity varies with conditions and analyte. Therefore, a calibration curve should be generated for each compound which is to be quantitated, and the samples should fall within the range of the calibration. Extrapolation is especially risky when the detector may be non-linear, and a large error may easily arise if the calibration line is extended beyond the last measured point.
Photoionization Detector (PID)
A detector which has much the same response as the FID, but which requires no support gases is the photoionization detector. This detector exposes the effluent stream to ultraviolet light, thus ionizing the sample. The ions are collected on an electrode, with the resulting current amplified and measured with an electrometer. Figure Chapter 4 .16 shows the detector. The range of compounds to which the detector is sensitive depends on the wavelength of the lamp used in the detector. Lamps can be purchased with wavelength peaks at 9.5, 10.0, 10.2, 10.7 and 11.7 eV. The 10.2 lamp is the most commonly used. The detector will respond to substances having ionization potentials below the lamp energy, and up to about 0.4 eV above it.
The PID is about 35 times more sensitive to aromatic compounds and somewhat more sensitive to alkanes than is the FID. It has a linear range of about 107. The response toward various compounds seems to be most closely related to their ionization potentials, with those with the lowest potentials giving the highest response. In general, as the carbon number increases, the sensitivity increases.
The chief advantages of the PID are that it is nondestructive, so it can be used in series with other detectors, and does not require support gases, as does the FID. This makes it ideal for portable instruments, which may use air for the carrier gas, and not require any cylinder gases to be carried. A portable PID detector, without a gas chromatograph, is available and can be used for screening for organic emissions, without speciation. The main drawback is a deposit which may form on the window separating the UV lamp from the gas stream. Some sample components react under the UV light and form solid products, which contaminate the lamp window.
Electrolytic Conductivity Detector (ElCD)
The electrolytic conductivity detector (Figure Chapter 4 .17) may be set up to determine compounds containing chlorine, sulfur or nitrogen. The detector is reconfigured, depending which of the elements are to be determined. When configured for chlorine, the effluent from the column is passed over a catalyst which converts any Cl to HCl. In sulfur mode, SO2 is formed, and in nitrogen mode, NH3. To reconfigure the detector, the catalyst is changed and reactor temperature is adjusted.
The reacted gas is scrubbed into a flowing aqueous or alcohol stream, and passed into a conductivity cell. The response of the detector is proportional to the number of Cl, N, or S atoms passing through the cell. The solvent is usually recycled through ion exchange resins. The detector must be watched for dips in the baseline which begin to occur as the solvent becomes exhausted. Detection limits of 10-12 g nitrogen/sec, 5 x 10-13 g chlorine/sec, and 10-12 g sulfur/sec are possible, and the selectivity ranges from 104 to 109 over hydrocarbons. The sensitivity is similar to that of the ECD, and the fact that the response is fairly consistent with the amount of the target heteroatom gives it an advantage over the ECD for some compounds.
Flame Photometric Detector(FPD)
This detector is designed for the specific detection of sulfur and phosphorous. It is similar in construction to the FID, but a cooler flame is produced by altering the hydrogen/air ratio. Instead of measuring the ions formed in the flame, the radiation emitted by the sulfur S2 and phosphorous HPO species formed when the sample components enter the flame is measured. A filter photometer is used to detect the radiation emitted at 394 nm for sulfur or 526 nm for phosphorous. Figure Chapter 4 .18 shows a schematic of this detector. Since the sulfur or phosphorous are measured in the same form for each component the response is governed by the total amount of the element in each sample component.
The detector is subject to negative interferences from hydrocarbons, which quench the emission if they are present in the flame at the same time as the sulfur or phosphorous compound being measured. A good GC separation will reduce this difficulty by eliminating the coelution of the interferent and the analyte.
The square root of the detector response is proportional to the sulfur concentration. For phosphorous, the response is directly proportional to concentration and is linear over 2-3 orders of magnitude.
Thermionic or Nitrogen-Phosphorous Detector (NPD)
The nitrogen-phosphorous detector is a modification of the flame ionization detector. In this detector a bead of a rubidium salt is placed at the tip of the flame. This gives a selectivity of 103 to 104 for N and P compounds over hydrocarbons. Helium carrier gas gives a better response with phosphorous compounds, while nitrogen is better for nitrogen containing compounds. The actual mechanism of the selective response is not entirely clear. It is believed that free radicals, formed in quantity by the nitrogen or phosphorous compounds in the flame, cause the vaporization and ionization of rubidium from the bead, adding to the signal. While the NPD shows selectivity toward the N and P-containing compounds the response to these is not a great deal higher than that of the unmodified FID. This detector gains its selectivity as much by repressing the response to hydrocarbons as by enhancing the response to N and P compounds. Its most common environmental use is in detection of nitro-PAH and other nitrogen-containing compounds in petroleum products.
Mass Selective Detector (MSD)
The mass selective detector, (MSD or GC/MS) is probably the most powerful tool in the hands of the environmental analyst. The detector, essentially a mass spectrometer, is a universal detector, as well as a very specific one. The MS can detect any molecule. Because each effluent component is fragmented and a mass spectrum is generated, plotting the intensity of a single mass fragment or group of fragments will generate a compound-specific chromatogram. These detectors will be discussed in Chapter 5.
Comparison of Detectors
The advantage of a universal detector is obvious, in that it responds to any component of the sample. Class specific detectors are also useful, since they can be used to simplify complex chromatograms. For most environmental work, the mass selective detector is the detector of choice, as it not only identifies but also quantitates the sample. It is mandated in many of the standard methods, but does not match the sensitivity of the ionization detectors. All the ions produced in an ionization detector are collected and measured, while each ion fragment in a mass spectrum is collected for only a small fraction of the time. This makes mass spectrometry inherently less efficient. However, one of the most difficult parts of the analysis with a general purpose detector is the identification of peaks. Because of the difficulty of reproducing the temperature exactly, and because the amount of loading on the column, especially of water vapor, may cause small shifts in retention time, retention times are not always sufficient to determine the identity of individual peaks unequivocally. A mass spectrum, combined with the retention time, is a much better way of making a reliable identification.
4.5 High Performance Liquid Chromatography
When a material is non-volatile or when it is so thermally fragile that it cannot be analyzed by gas chromatography, then liquid chromatography may be appropriate for the analysis. High performance columns are constructed of packing materials with very small particle diameters, on the order of 3-10 micrometers. Therefore, the eluent cannot flow through under gravity flow as was done in early liquid chromatographic analyses. A high pressure pump is necessary to force the eluent through the column at the rate which delivers the maximum number of theoretical plates. High pressure, high performance liquid chromatography is one of the essential tools of environmental analysis. It can readily handle high molecular weight compounds, such as polynuclear aromatic hydrocarbons, highly polar compounds such as phenols or organic acids, and even inorganic ions.
Choice of detector is somewhat limited in HPLC compared to GC. Since the eluent is usually an organic liquid, and ionization detectors, which are so useful in GC, are not applicable. Detection is therefore limited mostly to spectroscopic methods, which are limited by the absorbance characteristics of the analytes.
HPLC can be divided into several related techniques, depending on the separation mechanism and the column type. The most useful types in environmental analysis are reverse phase, normal phase and ion chromatography. Reverse phase liquid chromatography is probably the most frequently used, and the most versatile. It is called 'reverse' because of the comparison with 'normal phase', which is only called normal because it was invented first.
Reverse Phase Liquid Chromatography
Reverse phase columns have a packing composed of solid silica support particles which have an organic coating bonded to their surface. The bonded phase is produced by reacting a halogen substituted organosilane with the surface -OH groups present on the silica support. This leaves hydrocarbon chains, which may contain two, eight, or eighteen carbons, bonded at their ends through Si-O- Si groups to the surface of the support. Figure Chapter 4 .19 shows the bonding of octyl groups to the silica surface. These columns are designated by the carbon number of the chains attached, with the most frequently used column being the bonded octadecyl type, called C18.
Since these coatings are very non-polar in nature, the chief mechanism of retention is dispersion forces. This makes them useful for separation of organic compounds based on slight differences in their backbone or side chain configuration. The mobile phases commonly used are fairly polar in nature, with alcohols and water being common constituents. Since these are weaker eluents than the non-polar solvents which have a strong affinity for the highly non-polar column surface, sample components are retained long enough for good separation to take place. Components are eluted with the most polar ones being least retained and the least polar ones being held the longest.
Figure Chapter 4 .20 shows a chromatogram of a sample containing five compounds. In the first run, an isocratic eluent was used, a mixture of 30% methanol and 70% water. The first peaks are poorly separated while the later ones are too broad and take a long tome to elute. In the second case a gradient elution from 10% methanol to 100% methanol is done. The early peaks are better separated, since the initial eluent was weaker, while the late peaks are moved through the column more rapidly, as the eluent increases in strength. When gradients are done, it is important to begin with a weaker mobile phase, in this case one with a substantial amount of water. This allows the earliest peaks to remain in the column sufficiently long to achieve separation. Then the strength of the eluent is increased, by adding more of the less polar acetonitrile.
Gradients may be linear or curved. Figure Chapter 4 .21 shows some gradient profiles. If one had a situation in which several peaks were eluting close together at the beginning of the chromatogram, one might want to select a concave gradient similar to the one labeled C in the figure. This would allow the eluent to increase in strength very slowly, as these early peaks are being separated. Then, toward the end of the run, the strength is increased rapidly to bring out the later peaks. It is usually better to begin by experimenting with a linear gradient, then see if the beginning or end of the chromatogram could benefit from being stretched out a bit. A curved elution profile might then be tried.
Normal Phase Liquid Chromatography
Normal phase chromatography relies on such column packings as silica and alumina. Modern silica packings with polar bonded coatings are also available and are more reproducible and easy to use then is the bare silica. The difficulty with silica is its high affinity for water. Any trace of water in the solvent will be adsorbed onto the column, thereby changing its characteristics. This makes reproducible chromatography harder to achieve. Characteristically, normal phase columns have a polar surface, and eluents are rather non-polar, to achieve reasonable separation. In contrast to reverse phase separations, the strongest eluents used in this system are the most polar.
Solvent gradients, in this case, would begin with the least polar solvent, and gradually increase in polarity to bring out the most retained, most polar compounds. Samples best separated on normal phase columns are those comprised of different classes of compounds. Homologs are better separated on reverse phase columns.
4.6 HPlc Instrumentation
A complete apparatus consists of a pumping system, either for a single eluent or for a gradient, an injector, a column, one or more detectors and a data handling system. Typical setups are shown in Figure Chapter 4 .22.
Solvent Delivery Systems
Before being fed to the pumping system, the solvents should be filtered and degassed. Any particulate material in the solvent must be removed, because particulates may damage the pumps, and will, in time, collect at the top of the column and cause plugging. Degassing is important, because the dissolved gases may form bubbles when the pressure drops as the solvent enters the detector. Many detectors are severely disrupted by bubbles. Dissolved gases can be removed by purging the solvents with helium, which is quite insoluble in most solvents, or by passing the solvent through a microporous filter under vacuum. Vacuum filtration is the most common technique, since it accomplishes both the filtration and degassing processes at the same time. When solvents have been standing for some time, re-filtration is a good idea.
The characteristics of a HPLC pump which are of highest importance are its ability to deliver a constant, pulse-free flow, over a wide range of different flows. The materials of the pump system must be resistant to attack by the wide variety of mobile phases to be used. An additional desirable feature is the ability to generate a gradient of two or even three solvents, in a reproducible fashion. Pressures of up to 10000 psi are generated by the pump. Another consideration is the ease of changing solvents, which is related to the hold-up volume of the system.
Reciprocating dual piston pumping systems are the most common type. In these, one piston chamber is filling while the other is pumping. The pistons move in small chambers, each of which contains less than half a milliliter of eluent. A system of check valves keeps the solvent flowing in the correct direction. Pulses are kept to a minimum by elaborate design of the piston stroke cycle. As one piston begins to slow at the end of its stroke, the second one, newly filled, begins to deliver solvent, keeping the pressure and flow as constant as possible.
When using reciprocating piston pumps one must be careful to rinse the pump before turning it off. A solvent which contains no solids is pumped for several minutes, to remove any buffer salts which may remain from running an analysis. Salts can dry and crystallize on the surface of the piston when it is idle. Then, when the pump is restarted, the solids will cause abrasion of both the piston rods and the seals through which they pass.
A simpler, less expensive pumping system uses an eluent-filled syringe, driven by a constant speed motor, turning a screw drive. High pressure, pulseless flows can be generated by a syringe pump, but the volume is limited by the capacity of the syringe. This causes some downtime, as the pressure must be brought down, and the flow stopped, each time the syringe is refilled. These pumps are particularly useful for analyses using microbore columns which require very low flows. A constant pressure supplied by a compressed gas cylinder can also be used to drive a pump piston, giving a very smooth, pulseless flow. However, this system suffers from the same inconveniences as does the syringe pump.
Connections between solvent containers, pumps, columns and detectors are usually constructed of narrow bore stainless steel tubing. Extra-column band broadening is highly dependent on the radius of the tubing through which the sample and eluent pass. The smallest bore tubing which is practical without causing undue plugging should be used. Commonly, tubing around 0.01 inch i.d. is used. Increasing either the diameter or length of tubing through which the sample is passed will have a deleterious effect on the separation efficiency. The effect is more serious as smaller diameter columns are used.
When narrow-bore tubing is cut it is important to avoid plugging it, and to produce a smooth, square end for attachment of fittings. If the end of the tubing is cut raggedly or at a slant, a void space will usually occur when the tubing is placed into a compression fitting. Special cutting wheel tools are available to make good cuts.
Solvent Gradient Systems
Solvent gradient systems require a method of mixing a constantly changing amount of solvents from two or three separate reservoirs. The mixing may be done either before the high pressure pump, or after it. The low pressure mixing method uses low pressure metering pumps to deliver the components of the solvent mixture to the inlet of the high pressure pump. One must be careful to have no dissolved gases in the solvents because the gases may have lower solubility in the mixture, and bubbles can form.
High pressure gradient systems use separate high pressure pumps for each component, feeding into a small mixing chamber just before the injector. These are usually controlled by a microprocessor, which gradually increases the speed of one pump, while slowing the other. This is the most commonly used method, although it is somewhat more expensive, since it requires additional high pressure pumps. In addition to the ability to run samples which require gradient elution, another advantage of having a gradient system available is the ability to change the elution mixture easily. When setting up a new method, even one which will be done isocratically, it is faster and easier to make successive runs while having the microprocessor prepare different mixtures for trial runs, than it is to make the solvent mixtures by hand. In a laboratory with several instruments available, the gradient instrument should be used to set up methods, even for isocratic systems.
Sample Injectors
The requirement for an injector in HPLC is the same as it is for other types of chromatography. It must put a very narrow plug of sample into the eluent stream. One difficulty is that the eluent is under high pressure, which makes the use of a syringe impractical. The usual method is the use of sampling valve, containing a small sampling loop, with a volume of a few microliters. An excess of dissolved sample is flushed through the sampling loop, to fill it completely, with the excess passing out to waste. When the valve is rotated, the eluent flow is diverted through the loop, picking up the sample and moving it on to the column.
Some of these injection valves may also be used in a partial-filling method. In this case a volume of sample, smaller than that of the loop, is injected into the loop, using a syringe to measure the sample. This displaces some, but not all of the eluent in the loop. The accuracy is less than can be achieved by complete flushing of the loop, but it has the advantage that the sample volume can be readily adjusted. Figure Chapter 4 .23 shows a typical injection valve.
HPLC Columns
Precolumns and Guard Columns
The analytical column is expensive and can be damaged by particulate material depositing at the head of the column, as well as by attack of the eluent on the packing itself. Eluents with pH outside the 2 to 7 range may dissolve the silica support of the column. To lengthen the useful column life, guard and precolumns are used.
Precolumns are short segments of tubing packed roughly with similar material to that used in the column. The precolumn will pick up any particulates which are present at the exit of the pump. These particles may arise from poorly filtered eluents, or from wear fragments from the pump. More importantly, if the eluent is aggressive, and is dissolving the silica backbone of the packing, the precolumn serves to saturate the eluent with silica. Since it is located before the injector port, it cannot contribute to band broadening. Therefore, it is not necessary to have this column packed as carefully as an analytical column, or to be concerned about having very low dead volume fittings used to install it. It can be made of rather inexpensive components and packed in the lab.
Another source of particulate matter which may damage columns is the sample. To protect the column from materials in the sample which may deposit at the entrance of the column or which may be irreversibly adsorbed on the column packing, a guard column may be used. Guard columns can contribute to loss of separation efficiency because the sample passes through them. Therefore, they must be packed as carefully as the analytical column, and connected with low dead volume fittings. These columns are placed immediately before the analytical column, and can be considered to be the first part of the analytical column. Guard columns are commercially available, usually in 2-5 cm lengths. Some column systems are available which allow a replaceable cartridge to be placed in the inlet fitting of the analytical column, to serve as a guard column. Figure Chapter 4 .24 shows such a system.
Analytical Columns
Columns are usually constructed of stainless steel tubing with inner diameters of 4 or 5 mm. Microbore columns, with diameters of 1 and 2 mm are also available, as are columns with inside diameters above 10 mm, which are mainly used for preparative scale work. The end fittings on the column contain a frit to hold the packing in place, and a flow distributing plate which spreads the flow from the pump over the end of the column, thus helping to achieve constant flow through the entire cross section of the column.
Installation of columns is achieved by use of threaded
fittings, usually using a stainless steel compression ferrule. Many column
manufacturers use similar fittings, so that the same threaded nuts and fittings
can be used. However, once a steel ferrule is swaged onto a tube, it is often
not possible to move this tube to another fitting. The length of tubing which
protrudes from the ferrule may be different in each case. If the tubing is too
long, the ferrule cannot seat properly, and a good seal will not be achieved.
On the other hand, if the tube is too short, a gap will be present inside the
fitting and efficiency will be lowered. Figure Chapter 4 .25
shows this problem. Unless a system which uses a ferrule which can be
readjusted on the inlet or outlet tube is used, the ferrule should be cut off
and a new one fitted whenever columns are changed.
Eluents
The choice of eluent depends on the column and the sample. In reverse phase chromatography, a more polar eluent will move the sample slowly, and allow time for separation. A less polar solvent will elute late peaks more quickly and prevent excessive band broadening. There are several measures of eluent strength, including the polarity index, P’. A higher value of P’ indicates a more polar eluent. Often solvents are mixed to produce an eluent of a suitable strength for a particular separation. For instance, various mixtures of methanol and water are used to produce a variety of different polarities, with an increase in the water content making a less strong, more polar eluent for reverse phase work. The polarity index of a solvent mixture Pm composed of solvents ‘a’ and ‘b’ is computed as:
Pm = Pa * xa + Pb * xb ( Chapter 4 .20)
where Pa and Pb are the polarity indexes of a and b, and xa and xb is their volume fraction. The effect of eluent polarity on the capacity factor k’ of a compound is given by the equation:
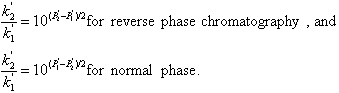 (
Chapter 4 .21)
(
Chapter 4 .21)
where P1’ and P2’ are the polarity indices of the two eluent mixtures.
The eluent must be able to keep the sample components in solution. The viscosity of the eluent is of concern, because a less viscous solvent can be used at a higher flow, without requiring very high pump pressures. Purity of the eluent, as well as its availability, cost and ease of disposal or recycling are other important considerations. Table Chapter 4 .5 lists some common eluent solvents and their physical characteristics important for HPLC.
Example:
In a reverse phase separation of a pesticide, the retention time was 15.5 min, with an eluent composed of methanol/water at a volume ratio of 30:70. An unretained peak eluted at 0.25 min. Calculate k’.
![]()
What water/methanol eluent composition will reduce k’ to 5?
Substituting values for methanol and water into Equation 4.20:
P’= 0.3 x 5.1 + 0.7 x 10.2 = 8.7
and ![]() so P2’
=6.52
so P2’
=6.52
To find the composition, let V = volume fraction of methanol.
6.52 = V x 5.51 + (1-V) x 10.2
V = 0.78. Therefore, the eluent is 78% methanol and 22% water.
4.7 HPLC Detectors
There is no sensitive universal detector available for use in HPLC. The only really universal, bulk property HPLC detector is the refractive index detector, which cannot be used with gradient elution, requires excellent temperature control, and is as much as 103 times less sensitive than other detectors. Therefore, it finds little use in environmental work. The detectors most often used are those such as absorption spectroscopic detectors, which respond to some property of the sample which is not exhibited by the mobile phase.
Ultraviolet Absorption Detectors
Ultra violet detectors are fairly general in application, since most organic compounds absorb some wavelengths in the UV spectrum. However, the spectral region of wavelengths below 210 is usually not useable for analysis because most solvents which would be used as eluents would also absorb in these areas. The response of this detector depends on Beer's Law, and therefore gives a linear response over four to five orders of magnitude. The detection limits vary widely, depending on the sample component and its extinction coefficient at the wavelength being used. In the most favorable cases, 1 ng or less of a compound may be detected.
Fixed wavelength detectors, using filters to isolate a single band of radiation, are inexpensive and stable. Light is passed through the filter then through a flow cell containing the effluent from the column. Finally, it is allowed to impinge on a photocell, where the light is measured. Generally, these are single beam instruments, but dual beam systems are possible. They lack versatility, since the only compounds which can be analyzed are those which absorb at the fixed wavelength. However, for standardized, repetitive analyses, these detectors may be ideal since their reproducibility is often slightly better than that of variable wavelength detectors.
Variable wavelength detectors, are, however, much more versatile. These use a continuum source and a monochromator to select the wavelength desired. A manually adjusted grating disperses the light and passes the target wavelength through the flow cell.
Detectors which can rapidly perform a complete scan over a range of wavelengths can give qualitative as well as quantitative information. This can be done with a rapid scanning instrument, but, more commonly a diode array detector is used. The photo diode array (PDA) detector uses an arrangement of diodes positioned so that each diode intercepts a different band of wavelengths. The signal from each diode is recorded, and a spectrum of the effluent at any moment is obtained. This is very useful in confirming identity of components, and even more, in determining the efficiency of separation. Figure Chapter 4 .26 shows the basic layout of a diode array detector.
The purity of a peak may be determined. Co-elution of components can be confirmed or ruled out by comparing spectra taken on the leading edge, the top, and the trailing edge of a peak. It is difficult to identify a totally unknown compound from the UV spectrum. These are relatively simple spectra, and the solvent, mixed with the sample, has an effect on the spectrum. However, comparison of samples and standards run in the same solvent, gives retention time and spectral information, and strong identification confirmation. Figure Chapter 4 .27 shows part of a chromatogram of a sample of polynuclear aromatic hydrocarbons run using a PDA. The peaks which have spectra and retention times matching those of the standard are identified, and the spectrum of each peak is printed on the report.
The flow cells used for absorption detectors are designed to give the maximum length of sample for the light to pass through, while keeping the volume as small as possible, to insure that the resolution is not compromised. Figure Chapter 4 .28 shows a UV detector cell. The cell has a Z shape to provide the maximum path length with as little cell volume as possible. The principal source of noise in absorbance detectors using a flowing sample is due to slight changes in refractive index. These are due to slight inhomogeneities in the composition of the eluent, changes in temperature, or turbulence in the flow. The change in refractive index diverts some of the light from the path to the detector, momentarily decreasing the signal.
Fluorescence Detectors
Fluorescence detectors are among the most sensitive available. These are most suitable for, but are not limited to, compounds which fluoresce. Non-fluorescent compounds may be derivatized by adding a reagent after the column, which supplies a fluorescent tag to the sample molecules. Alternatively, the eluent may be made fluorescent and the sample peaks detected by the decrease of fluorescence as the peak elutes. This is known as "vacancy chromatography".
Fluorescence detectors need an intense high energy source, either line or continuous, to excite the fluorescence. Mercury lamps are used for line source excitation, and deuterium or xenon arc sources for continuum source. A monochromator is used to select the wavelength for excitation and for emission. The wavelength selection can also be done with filters, at the expense of versatility and sensitivity. The wavelength at the absorbance peak may not be available in a filter instrument, so that the highest sensitivity cannot be achieved. An photomultiplier is used to capture and amplify the weak emission from the fluorescent molecules. Figure Chapter 4 .29 shows a fluorescence detector.
For dilute solutions the equation which relates the emission to the concentration is:
If = Io ff (2.3 abC) ( Chapter 4 .22)
where: If is the measured emission intensity, Io is the excitation beam intensity, ff is the number of photons emitted per photon absorbed (the quantum yield), a is the molar absorption coefficient, b is the cell path length, and C is the sample concentration. The response is linear over about two orders of magnitude. Sensitivity varies widely, depending on the amount of light scattering in the optical system, the intensity of the excitation radiation, and the fluorescence quantum efficiency of the sample. Mobile phase composition is also important since fluorescence is readily quenched. Oxygen is a particularly efficient fluorescence quencher, so solvents must be well degassed. Fluorescence is also temperature dependent, and, at higher concentration, self absorbance can be serious.
Mass Spectrometric Detection
The most informative detector is probably the mass spectrometer. Interfacing between the HPLC and the ion source is even more difficult than it is with GC/MS. The eluent is a liquid, and therefore, must be eliminated in some way before the sample is injected into the vacuum system.
4.8 Ion Chromatography
Ion chromatography is used for separation of ionic species. The stationary phase is an ion exchange resin, and retention of the ionic species occurs as they are exchanged onto and off the resin surface. A cation exchange resin has R-H+ groups on its surface and, a cation such as Zn++ is retained because it exchanges with the hydrogen ions on the resin:
R-H+ + Zn++ Û R-Zn++ + H+
Similarly, an anion exchange resin R-OH-, will exchange OH- ions for anions such as NO3- in the sample:
R-OH- + NO3- Û R-NO3- + OH-
The partition coefficient K for the cation exchange is:
K = [R-Zn++]/[Zn++]
where R-Zn++ is the concentration on the ion exchange resin, and Zn++ is the concentration in the mobile phase. The partition coefficient for the anion exchange is calculated in a similar fashion. The most common anion exchange column incorporates a quaternary amine group, while cation columns usually bear sulfonate groups. The packings are prepared by sulfonating or aminating the surface of the polymer core, with the active sites located close to the surface, to improve the mass transfer between the eluent and the stationary phase.
The instrumentation used for ion chromatography is similar to that used for HPLC and is shown in Figure Chapter 4 .30. The conductivity detector which measures the electrical conductivity of the eluting mobile phase is commonly used.
If the mobile phase has a high ionic strength, the background electrical conductivity will be high and the detector will have low sensitivity. There are two methods used to reduce this difficulty: the suppresser column technique and the single column technique. In the suppresser column method, the a fairly strong eluent is used to move the sample through the analytical column. Then the eluent is passed through a second column, the suppresser column. This neutralizes the eluent and allows easy detection of the sample ions. For instance, in the analysis of cations, a dilute HCl solution may be used as the eluent. The analytical column is a low capacity cation exchange resin, and the suppresser column is a high capacity anion exchange resin. The large excess of H+ ions displaces the sample cations, with each cation establishing its own equilibrium between the eluent and the surface. The suppresser column is an anion exchange resin in the hydroxyl form, and the H+ ions from the mobile phase react with the OH-, forming water. This leaves the sample cations in the eluent stream, with a very low background conductivity, facilitating conductivity detection. The suppresser column eventually becomes exhausted and must be regenerated to replenish the OH- on the surface.
For analysis of anions, the analytical column is an anion exchange resin while the suppresser is a high capacity cation exchange resin. The eluent is, for instance, a dilute solution of NaOH. In the suppresser column, the OH- ions in the eluent are neutralized by the H+ from the column. This leaves only the sample anions in the solution and high sensitivity is obtained.
The single column method, a more recent development, uses a low capacity ion exchange resin designed especially for chromatographic purposes. Since the resin has such low retention, the eluents of very low ionic strength can be used. Buffers of such weak acids as boric acid have very low conductivities and the detection of the sample ions can be done without the use of a suppresser column. This simplifies the system, and allows the usual HPLC equipment, with only a conductivity detector added, to be applied to ion chromatography.
Ion chromatography can be used for the detection and quantitation of many species: Inorganic anions such as chloride, fluoride, sulfate, nitrate and nitrite; cations such as sodium, calcium, copper, lead, ammonium ions; as well as ionizable organic species such as carboxylic acids and amino acids can be determined using this technique. While many metals may be more easily determined by atomic spectroscopy, ion chromatography has the ability to distinguish between species having different oxidation states, such as Fe(II) and Fe(III). This is not possible if atomic absorption spectroscopy is used for the analysis. Figure Chapter 4 .31 shows an ion chromatogram of an extract of anions from an air filter, obtained with a single column system.
4.9 Supercritical Fluid Chromatography
A substance cannot exist in the liquid state at a temperature above its critical temperature. However, if a material is above its critical temperature, and is subjected to sufficiently high pressure, it becomes much more dense than ordinary gases, and takes on some liquid-like properties. This is then referred to as a supercritical fluid. Figure Chapter 4 .32 shows the phase diagram for CO2, a commonly used supercritical fluid. The properties of supercritical fluids can be continuously varied between those of the gas and those of the liquid by changing the temperature and pressure. These fluids can be used as mobile phases in chromatography. Properties which can be varied include the viscosity, solvent properties and diffusivity, all of which are important chromatographic properties.
The properties of these fluids are usually closer to those of liquids than gases. The solubilizing power of a supercritical fluid is much greater than that of a gas. Therefore, nonvolatile and slightly volatile compounds may be separated by supercritical chromatography, while this would be impossible to do with GC. There is also an advantage over HPLC analysis for these compounds, since the solute diffusion coefficients in supercritical fluids are much greater. This means that the eluent velocity required for the maximum column efficiency is 5 to 10 times greater than that for HPLC. Equally efficient separations can therefore be done in much less time than is needed for HPLC. Finally, the viscosity of these fluids is much lower than that of liquids, making them much easier to pump through columns at a faster flow. Both packed and open tubular columns are used.
Any substance stable above its supercritical temperature might be used for eluent in SFC, but only a few are used routinely. Supercritical fluids which have been used are carbon dioxide, nitrous oxide, sulfur hexafluoride, Freon-13, ethane and ammonia. Of these, CO2 is the most common, since its critical temperature, 31oC, is readily attained, it is non-toxic, and is readily available. Between the pressures of 72 and 400 atmospheres, and temperatures of 40 to 140 oC, the density of CO2 can be varied from 0.1 to almost 1 mg/ml. The only practical supercritical fluid which is reasonably polar is ammonia. It is, however, quite reactive and difficult to use. Modifiers are therefore used to improve the separation of more polar substances in nonpolar supercritical fluids such as CO2. Modifiers are added to the eluent to improve peak shape and shorten retention times. These modifiers, including methanol, water and formic acid, are used in low concentrations, below 2% by volume. Modifiers at this low level may be thought of as deactivating silanol groups on the column, rather than increasing the solubility of the sample compounds in the solvent.
Programming in SFC is quite flexible, since temperature, pressure and density all affect the retention of samples. The most common method of programming is density or pressure programming although temperature programming has also been used.
SFC Instrumentation
The components of an SFC system are similar to those of HPLC, since a pump is used to produce the high pressures required, but GC and HPLC detectors can be used. A typical system is shown in Figure Chapter 4 .33.
A syringe pump is the most commonly used pump although reciprocating pumps have been used for packed column work. The mixing of modifiers complicates the system. Cylinders of eluent with modifier already mixed may be purchased, but then the amount of modifier cannot be adjusted. A second pump to add modifier is useful.
Samples are injected into the system using high pressure rotary sampling valves. Sample volumes as small as a few nanoliters are needed for capillary work, so sample splitting techniques may be required. However, the sample is injected at room temperature, where the eluent may not be supercritical. The sample also may not be homogeneously mixed into the eluent quickly enough before the split is made. Therefore, the operation of a splitter is not always simple, and quantitation may be poor. Sample volumes for packed columns are larger, in the microliter range, so injection is a much easier task.
Columns used in SFC are usually small bore columns packed with 5 to 10 mm bonded phase particles similar to those used for HPLC. Short capillary columns of 1 to 10 meters in length are also used, with stationary phases which are often more crosslinked than those used in GC. All bonded stationary phases have some contribution from unreacted silanol groups, and these can be a problem in SFC, because of the relatively nonpolar nature of CO2. This is why polar modifiers are effective.
At the end of the column a restrictor is required to keep the fluid in the column at the required pressure. The restrictor is positioned before the detector when the detector is a GC type detector, and after the detector, when an HPLC type detector such as a UV absorption cell, is used. A restrictor may be simply a short length of narrow bore fused silica tubing of 5 to 15 microns i.d. However, the sample may precipitate as fog droplets when the solvent suddenly decompresses at the end of the restrictor. These droplets, when fed into a flame ionization detector, cause signal spikes. This may be avoided by decompressing the eluent more gradually in a tapered or conical restrictor, which is kept warm at the tip so that the sample has a chance to evaporate.
The flame ionization detector is probably the most common detector for SFC, as it is compatible with the usual fluids. While organic modifiers interfere, water and formic acid can be used with FID detectors. The sensitivity of the FID is somewhat lower than its GC counterpart. The UV detector is also often used, especially when wide bore columns or organic modifiers make the FID unsuitable. The volume of the absorbance cell must be very small in SFC, on the order of 50 nanoliters or less, to avoid band broadening when capillary columns are used. Standard HPLC UV cells may be used in packed column work, but modifications may be necessary to allow the cells to be used at the much higher pressures common in SFC.
4.10 Applications of Chromatography in Environmental Analysis
Gas chromatography is the most widely used separation technique. Many of the target pollutants are volatile enough to be analyzed by GC. For semivolatiles such as PAH, PCBs and some pesticides HPLC is widely used. GC has several advantages over HPLC. GC columns provide a larger number of plates and a variety of highly sensitive and selective detectors are available. Since environmental samples are complex, the high separation capability is very important. If there is a choice between GC and HPLC, GC is usually preferred. An example of a difficult separation of some metabolites of benzo-a-pyrene, too non-volatile to be separated easily by GC, is shown in Figure Chapter 4 .34.
Supercritical chromatography has still not become a standard technique in environmental analysis. Most samples can be done by either GC or HPLC, both of which are much more mature techniques. There are some advantages, especially the ability to use high sensitivity GC detectors for relatively nonvolatile samples, but the equipment has not yet reached the sophistication and ease of use of GC or HPLC. Supercritical extraction, on the other hand, will probably develop into a major technique for environmental sample preparation, because it readily replaces toxic solvents.
References and Suggested Readings
1.
Principles
of Instrumental Analysis; D.A. Skoog and J. J. Leary, Saunders College
Publishing, New York, 1992.
2.
Instrumental
Methods of Analysis; H. H. Willard et. al.; 6th edition, Wadsworth.
3.
Gas
Chromatographic Environmental Analysis; Fabrizio Bruner, VCH Publishers, Inc,
New York, 1993.
4.
Chromatography
Today, C.F. Poole and S.K.Poole, Elsevier, 1991
5.
Ion
Chromatography; J. Weiss, 2nd Ed., 1995, VCH Publishers, New York.
6.
Introduction
to Modern Liquid Chromatography; L. R. Snyder and J. J. Kirkland, 2nd Ed. 1980,
John Wiley and Sons, New York.
7.
Gas
Chromatographic Environmental Analysis: Principles, Techniques and
Instrumentation, F. Bruner, VCH Publishers, New York, 1993
Study Questions
1.
What
are the common factors which exist in all types of chromatography?
2.
What
is the significance of to in gas chromatography?
3.
A
gas chromatographic peak from a 25 m long column has an adjusted retention time
of 11.2 minutes, and a width at half height of 20 seconds. How many theoretical
plates are present in the column? What is the value of H?
4.
A
sample of pesticide is analyzed by gas chromatography. A 0.1 ml injection of a standard containing 0.234 mg/l gives a peak of area 34873. The same size
injection of an unknown sample solution gives an area of 39945. What is the
concentration of the pesticide in the sample solution?
5.
In
gas chromatography predict the effect on the efficiency of a separation when
the following changes are made:
a. Particle size of the packing is increased
b. Gas flow is increased
c. Thickness of liquid phase in a capillary column is increased
6. In an HPLC analysis, a reverse phase column is being used with a solvent gradient, starting with solvent A and increasing concentration of solvent B over time. The program starts with 50% A and 50% B and runs linearly, reaching 100%B at 20 minutes. Explain which of the two solvents is more polar.
7. What are some of the properties of a good chromatographic detector.
8. What is the advantage in using a halogen specific detector such as the ECD in environmental analyses?
9. Define the following terms:
Gas-Liquid chromatography.
Bonded phase
Normal phase HPLC
Gradient elution
10. What is the function of the suppresser column in ion chromatography? Under what conditions is it not needed?
11. Predict the
elution order of the following in reverse phase and in normal phase
chromatography: (a) n-heptane, heptanol, and toluene.
(b)
nitrobenzene, benzene and phenol
12. What parameters can be used to improve resolution in GC? in HPLC?
13. What are some advantages of SFC over GC? Over HPLC?
14. In a normal phase LC separation, nitrobenzene had a retention time of 28.0 min, while an unretained compound eluted in 0.9 min. The mobile phase was a 50:50 mixture of chloroform and hexane. What mixture of chloroform and hexane will reduce the k’ to 9.0.
The following data were obtained for a separation using a 0.5 mm id, 10 meter open tubular column:
|
|
Retention time (min) |
Peak base width (min) |
|
Methane |
2.0 |
|
|
Cyclohexane |
8.0 |
0.65 |
|
Methylcyclohexane |
8.17 |
0.72 |
|
Toluene |
10.2 |
0.95 |
Draw the chromatogram and label the peaks
b) Calculate the number of peaks for each compound
c) Calculate the capacity factor for toluene
d) Calculate the length of the column needed to separate cyclohexane and methylcyclohexane at a resolution of 1.6.
15) In a reverse phase separation of chlorinated phenols, trichlorophenol has a retention time of 15.0 min, when 30:70 acetonitrile-water mixture is used as the mobile phase. What mixture of these two solvents will reduce the retention time to 12.0.