Updown Distance: A New Distance Measure
for Comparing Two Phylogenetic Trees
Jason T. L. Wang
Bioinformatics Program
Department of Computer Science
New Jersey Institute
of Technology
wangj@njit.edu
Steven Regula
Department of Computer Science
New Jersey Institute of Technology
Dennis Shasha
Courant Institute of Mathematical Sciences
Department of Computer Science
New York University
Lei Hua
Department of Computer Science
New Jersey Institute of Technology
Rekha Patel
Department of Computer Science
New Jersey Institute of Technology
Introduction
We describe an efficient method for evaluating closeness of tree matches when comparing a query tree P with a data tree D where both P and D are phylogenetic trees. The method computes the Updown distance between P and D.
The phylogenetic trees satisfy the following properties:
For more information and examples of computing the Updown distance of phylogenetic trees with this tool, see Example 1, Example 2, Example 3, or Example 4 and also some highlighted Special Cases.
Installation
This phylogenetic tree matching algorithm has been implemented in Java. As such, all that is required is a Java virtual machine, version 5 or greater to execute the software, and optionally, the SDK version 5 or later to modify and compile. You can download the latest version of Java from Sun Microsystems at: http://java.sun.com/javase/downloads/index.jsp.
To download only the class files and execute:
1. First create a folder called "phylogeny".
2. Second, store the following class files in the folder you just created: UpdownDistance.class, UpdownDistance$Node.class.
3. To execute, from the directory above phylogeny, type: "java phylogeny/UpdownDistance".
To download the source, compile, and execute:
1. Create a folder called "phylogeny".
2. Download and save the following source file: "UpdownDistance.java " to the folder you just created.
3. Compile by typing "javac phylogeny/UpdownDistance.java" from the directory above "phylogeny". You need to have Java SDK 5 or greater installed to eliminate the risk of Java incompatibilities.
4. To execute, from the directory above phylogeny, type: "java phylogeny/UpdownDistance".
Documentation:
Code documentation is available online at: Javadoc. An Internet Explorer version 7 or a later release is needed to browse the code documentation.
Usage
This software is intended to be widely available and as such has been implemented using Java. To further facilitate widespread experimentation with this software, the trees are input using the standard "Newick" format. For more information about the Newick format, see the Wikipedia entry and also a graphical tree generation tool by using the "Links" area at the bottom of this document. Below is a sample execution of the program comparing the trees in (i) and (ii). Note that in (ii) the data tree D has been marked before final reduction. Following the example execution is a more in depth detailing of all input and output.
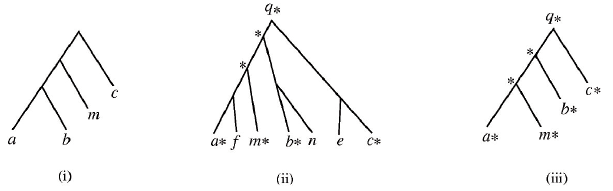
(i) is an example of a query tree P.
(ii) shows a marked data tree D; marking is an important step in reduction.
(iii) shows the reduced data tree D*, which will be compared to P to complete the matching algorithm.
In general, reduction works as follows:
Consider the query tree P and a data tree D. Find the nodes that occur in both P and D. Mark those matching nodes in D with asterisks. If two distinct nodes of D are marked, then their least common ancestor is also marked. We then consider the reduced data tree D* of D that contains only the marked nodes.
For a complete description of reduced tree and the reduction process, please see pages 39-40 of the published document listed under Citation.
Complete Execution:
ocean Updown_distance>: java phylogeny/UpdownDistance
Enter your query
tree: (((a,b),m),c);
Enter your data tree:
((((a,f),m),(b,n)),(e,c))q;
The query tree is: (((a,b),m),c);
The data tree is: ((((a,f),m),(b,n)),(e,c))q;
The reduced data tree is: (((a*,m*)*,b*)*,c*)q*;
Updown Distance: 6
Input
The only input required for successful execution of the system are two valid trees in the Newick format. The first tree the system prompts for is the query tree P. Be sure to end the case sensitive Newick formatted string with a semicolon.
The example entered in the execution above was: (((a,b),m),c);
The second input prompt was for the data tree that the system will be matching against.
In this example the data tree D was: ((((a,f),m),(b,n)),(e,c))q;
Output
Once the user has successfully entered both the query and data trees using the Newick format, the system begins to process the trees, execute the matching algorithm, and print results.
The first items delivered to the console are "confirmation" trees printed back to the user. By design, the strings delivered back to the user should look exactly like the ones entered in the previous step. Note, however, that they are not simply static strings sent back to the user but rather generated and displayed from the tree Node class itself. If these strings were to differ from their corresponding input strings, it would be likely that some parsing error had occurred. Below is an example of this output step:
The query tree is: (((a,b),m),c);
The data tree is :
((((a,f),m),(b,n)),(e,c))q;
The second section of output from this system is a rendering of what the reduced data tree D* would look like. Also note that the reduced data tree is represented in Newick format as well; however asterisks mark nodes that have been preserved during reduction. Nodes without asterisks are not considered during the tree matching algorithm. Below is an example of this output:
The reduced data tree is:(((a*,m*)*,b*)*,c*)q*;
The third piece of output is the Updown distance of the two trees, which should always be an integer value. The following is an example of this output:
Updown Distance: 6
Input File Option
This section highlights some basic details for using input files; for more in depth information, see the tutorial.
The example above shows the system executing in interactive mode. When a user launches the program without any command line parameters, the system prompts for Newick formatted query and data tree strings. However, there are platform dependent limitations on input string length that could otherwise impact a user's ability to compare large phylogenetic trees. To elucidate this detail, the following chart highlights the maximum interactively entered input string length among various platforms:
|
Platform Tested |
Maximum Newick Formatted Tree Length |
| SunOS 5.9 (UNIX) | 258 |
| Apple OSX | 1025 |
| Linux Fedora Core 4 | 4096 |
| MS Windows Vista | 8191 |
| MS Windows XP | 7658 |
Note: The terminology "Newick Formatted Tree Length" represents the number of characters that comprise the string representation of the input tree in Newick format. For example, if a query string of "((a,b)c)" is entered, its corresponding length would be 8 characters.
To overcome command line interface input string length limitations, the system allows for parameters that specify filenames of Newick formatted input strings. To use this option, first create and save a plain text file using a text editor. A sample Newick formatted tree in the MS Windows "notepad" application is represented below:
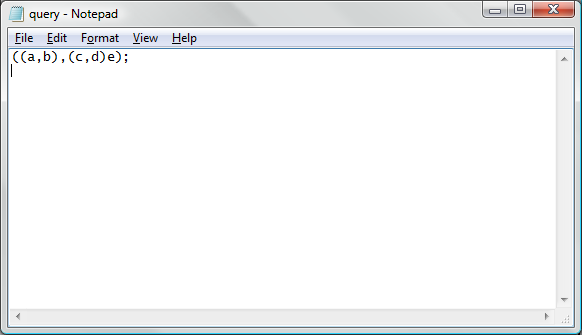
Note that in this image, there is a new line after the Newick formatted tree. After the input file has been generated, it can be used for processing using the following command line parameters.
java phylogeny/UpdownDistance /?
Usage: java UpdownDistance [-p QUERYFILENAME] [-d DATAFILENAME]
Therefore, to use the updown distance calculator with datafiles as input, the user would type the following if the query and data trees were named query.txt and data.txt, respectively.
java phylogeny/UpdownDistance -p query.txt -d data.txt
An example is output from the launch initiated above:
Reading query tree file.
Reading data tree
file.
The query tree is: ((a,b),(c,d)e);
The data tree is: ((a,b,c)e,(z,y)d)g;
The reduced data tree is: ((a*,b*,c*)e*,d*)g*;
Updown Distance: 12
Special Notes on Newick Formatted Tree String Size Limits:
As the underlying technology for interpreting Newick formatted trees is the Java String class, this system imposes no restriction on the length of the Newick formatted tree string when stored in a file. However, users should remember that there are finite limits to computer memory systems and will thus encounter overflow upon reaching the limit of the host system. For a brief but interesting read on Java String limitations, see: What is the largest text file that Java can read into a String?
Citation
Jason T. L. Wang, Huiyuan Shan, Dennis Shasha and William H. Piel, "Fast Structural Search in Phylogenetic Databases," Evolutionary Bioinformatics Online, Volume 1, 2005, pp. 37-46.
Links
Newick Notation - Wikipedia Entry
Phyfi - Online tool for drawing and manipulating phylogeny color figures
What is the largest text file that Java can read into a String?
Download Issues
Some browsers open the PDF file and the Web page manuals and programs instead of starting a download. If this happens, try right-clicking on the link and choosing an option named "Save Target As..." or similar. If a separate window is popped up, click "File" on the top bar menu of the window and click on "Save As" to save the file.